Differential Expression
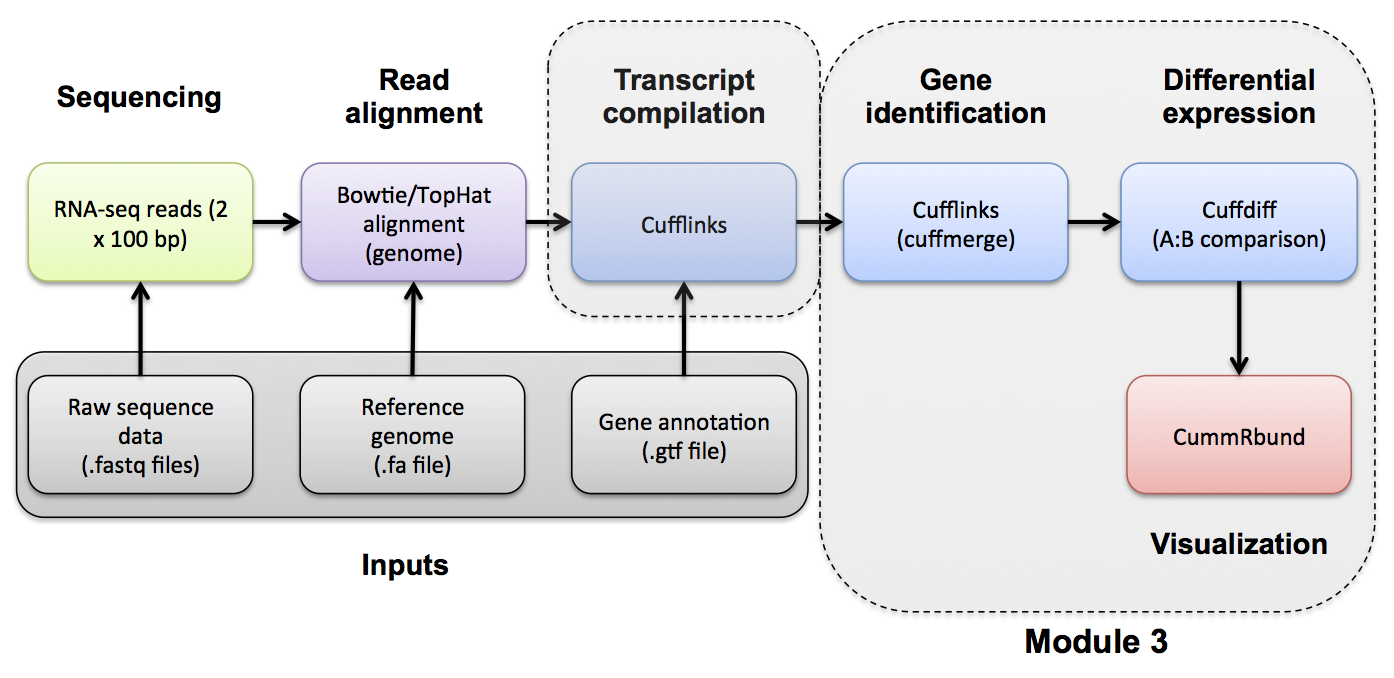
#3-ii. Differential Expression Use Cuffmerge and Cuffdiff to compare the tumor and normal conditions. Refer to the Cufflinks manual for a more detailed explanation:
- http://cole-trapnell-lab.github.io/cufflinks/cuffmerge/index.html
- http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/index.html
Cuffmerge basic usage:
cuffmerge [options]* <assembly_GTF_list.txt>
- "assembly_GTF_list.txt" is a text file "manifest" with a list (one per line) of GTF files that you would like to merge together into a single GTF file.
Extra options specified below:
- '-p 8' tells cuffmerge to use eight CPUs
- '-o' tells cuffmerge to write output to a particular directory
- '-g' tells cuffmerge where to find reference gene annotations. It will use these annotations to gracefully merge novel isoforms (for de novo runs) and known isoforms and maximize overall assembly quality.
- '-s' tells cuffmerge where to find the reference genome files
Merge all 6 cufflinks results so that they will have the same set of transcripts for comparison purposes
cd $RNA_HOME/expression/tophat_cufflinks/ref_only/
ls -1 *Rep*ERCC*/transcripts.gtf > assembly_GTF_list.txt
cuffmerge -p 8 -o merged -g $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf -s $RNA_HOME/refs/hg19/bwt/chr22_ERCC92/ assembly_GTF_list.txt
###OPTIONAL ALTERNATIVE - Merge Cufflinks GTFs for STAR Perform the merge step for STAR-alignment-based cufflinks output:
cd $RNA_HOME/expression/star_cufflinks/ref_only/
ls -1 *Rep*ERCC*/transcripts.gtf > assembly_GTF_list.txt
cuffmerge -p 8 -o merged -g $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf -s $RNA_HOME/refs/hg19/fasta/chr22_ERCC92/ assembly_GTF_list.txt
###OPTIONAL ALTERNATIVE - Merge Cufflinks GTFs for HISAT2 Perform the merge step for HISAT2-alignment-based cufflinks output:
cd $RNA_HOME/expression/hisat2_cufflinks/ref_only/
ls -1 *Rep*ERCC*/transcripts.gtf > assembly_GTF_list.txt
cuffmerge -p 8 -o merged -g $RNA_HOME/refs/hg19/genes/genes_chr22_ERCC92.gtf -s $RNA_HOME/refs/hg19/fasta/chr22_ERCC92/ assembly_GTF_list.txt
Cuffdiff basic usage:
cuffdiff [options] <transcripts.gtf> <sample1_hits.sam> <sample2_hits.sam> [... sampleN_hits.sam]
- Supply replicate SAMs as comma separated lists for each condition:
- Example: sample1_rep1.sam,sample1_rep2.sam,...sample1_repM.sam
- '-p 8' tells cuffdiff to use eight CPUs
- '-L' tells cuffdiff the labels to use for samples
Create necessary directories:
cd $RNA_HOME/
mkdir -p de/tophat_cufflinks/ref_only
cd $RNA_HOME/alignments/tophat/
Generate the cuffquant binary format files for cuffdiff
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep1_ERCC-Mix1 $RNA_HOME/expression/tophat_cufflinks/ref_only/merged/merged.gtf UHR_Rep1_ERCC-Mix1/accepted_hits.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep2_ERCC-Mix1 $RNA_HOME/expression/tophat_cufflinks/ref_only/merged/merged.gtf UHR_Rep2_ERCC-Mix1/accepted_hits.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep3_ERCC-Mix1 $RNA_HOME/expression/tophat_cufflinks/ref_only/merged/merged.gtf UHR_Rep3_ERCC-Mix1/accepted_hits.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep1_ERCC-Mix2 $RNA_HOME/expression/tophat_cufflinks/ref_only/merged/merged.gtf HBR_Rep1_ERCC-Mix2/accepted_hits.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep2_ERCC-Mix2 $RNA_HOME/expression/tophat_cufflinks/ref_only/merged/merged.gtf HBR_Rep2_ERCC-Mix2/accepted_hits.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep3_ERCC-Mix2 $RNA_HOME/expression/tophat_cufflinks/ref_only/merged/merged.gtf HBR_Rep3_ERCC-Mix2/accepted_hits.bam
Perform UHR vs. HBR comparison, using all replicates, for known (reference only mode) transcripts:
cuffdiff -p 8 -L UHR,HBR -o $RNA_HOME/de/tophat_cufflinks/ref_only/ --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/expression/tophat_cufflinks/ref_only/merged/merged.gtf UHR_Rep1_ERCC-Mix1/abundances.cxb,UHR_Rep2_ERCC-Mix1/abundances.cxb,UHR_Rep3_ERCC-Mix1/abundances.cxb HBR_Rep1_ERCC-Mix2/abundances.cxb,HBR_Rep2_ERCC-Mix2/abundances.cxb,HBR_Rep3_ERCC-Mix2/abundances.cxb
###OPTIONAL ALTERNATIVE - CuffDiff for STAR results perform the cuffdiff step for STAR-alignment-based cuffmerge output:
cd $RNA_HOME/
mkdir -p de/star_cufflinks/ref_only
cd $RNA_HOME/alignments/star/
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep1 $RNA_HOME/expression/star_cufflinks/ref_only/merged/merged.gtf UHR_Rep1/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep2 $RNA_HOME/expression/star_cufflinks/ref_only/merged/merged.gtf UHR_Rep2/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep3 $RNA_HOME/expression/star_cufflinks/ref_only/merged/merged.gtf UHR_Rep3/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep1 $RNA_HOME/expression/star_cufflinks/ref_only/merged/merged.gtf HBR_Rep1/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep2 $RNA_HOME/expression/star_cufflinks/ref_only/merged/merged.gtf HBR_Rep2/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep3 $RNA_HOME/expression/star_cufflinks/ref_only/merged/merged.gtf HBR_Rep3/Aligned.out.sorted.bam
cuffdiff -p 8 -L UHR,HBR -o $RNA_HOME/de/star_cufflinks/ref_only/ --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/expression/star_cufflinks/ref_only/merged/merged.gtf UHR_Rep1/abundances.cxb,UHR_Rep2/abundances.cxb,UHR_Rep3/abundances.cxb HBR_Rep1/abundances.cxb,HBR_Rep2/abundances.cxb,HBR_Rep3/abundances.cxb
###OPTIONAL ALTERNATIVE - CuffDiff for HISAT2 results perform the cuffdiff step for HISAT2-alignment-based cuffmerge output:
cd $RNA_HOME/
mkdir -p de/hisat2_cufflinks/ref_only
cd $RNA_HOME/alignments/hisat2/
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep1 $RNA_HOME/expression/hisat2_cufflinks/ref_only/merged/merged.gtf UHR_Rep1/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep2 $RNA_HOME/expression/hisat2_cufflinks/ref_only/merged/merged.gtf UHR_Rep2/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o UHR_Rep3 $RNA_HOME/expression/hisat2_cufflinks/ref_only/merged/merged.gtf UHR_Rep3/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep1 $RNA_HOME/expression/hisat2_cufflinks/ref_only/merged/merged.gtf HBR_Rep1/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep2 $RNA_HOME/expression/hisat2_cufflinks/ref_only/merged/merged.gtf HBR_Rep2/Aligned.out.sorted.bam
cuffquant -p 8 --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check -o HBR_Rep3 $RNA_HOME/expression/hisat2_cufflinks/ref_only/merged/merged.gtf HBR_Rep3/Aligned.out.sorted.bam
cuffdiff -p 8 -L UHR,HBR -o $RNA_HOME/de/hisat2_cufflinks/ref_only/ --library-type fr-firststrand --frag-len-mean 262 --frag-len-std-dev 80 --no-update-check $RNA_HOME/expression/hisat2_cufflinks/ref_only/merged/merged.gtf UHR_Rep1/abundances.cxb,UHR_Rep2/abundances.cxb,UHR_Rep3/abundances.cxb HBR_Rep1/abundances.cxb,HBR_Rep2/abundances.cxb,HBR_Rep3/abundances.cxb
What does the raw output from Cuffdiff look like?
cd $RNA_HOME/de/tophat_cufflinks/ref_only
ls -l
head isoform_exp.diff
grep -P "gene_id|OK" isoform_exp.diff | cut -f 2-6,8-10,12 | sort -k 9,9 | less -S
Press 'q' to exit the 'less' display
How many genes are there on this chromosome?
grep -v gene_id gene_exp.diff | wc -l
How many were detected above 0 in UHR or HBR (take the sum of expression values for both and check for greater than 0)?
grep -v gene_id gene_exp.diff | perl -ne '@line=split("\t", $_); $sum=$line[7]+$line[8]; if ($sum > 0){print "$sum\n";}' | wc -l
How many differentially expressed genes were found on this chromosome (p-value < 0.05)?
grep -v gene_id gene_exp.diff | cut -f 12 | perl -ne 'if ($_ < 0.05){print "$_"}' | wc -l
Display the top 20 DE genes. Look at some of those genes in IGV - do they make sense?
grep -P "OK|gene_id" gene_exp.diff | sort -k 12n,12n | head -n 20 | cut -f 3,5,6,8,9,10,12,13,14
Save all genes with P<0.05 to a new file.
grep -P "OK|gene_id" gene_exp.diff | sort -k 12n,12n | cut -f 3,5,6,8,9,10,12,13,14 | perl -ne '@data=split("\t", $_); if ($data[6]<=0.05){print;}' > DE_genes.txt
| Previous Section | This Section | Next Section | |:-------------------------------:|:---------------------------------------------------:|:-----------------------------------------:| | Expression | Differential Expression | DE Visualization |