Reference Guided Transcript Assembly
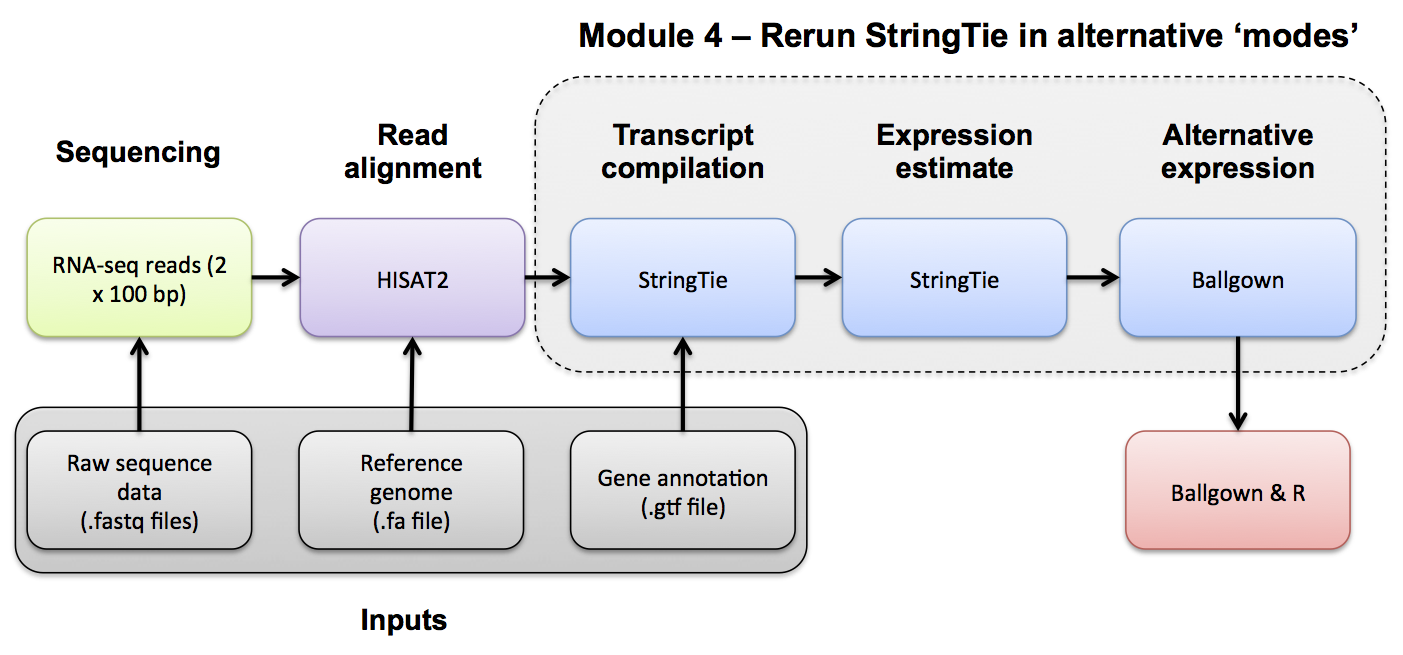
Note on de novo transcript discovery and differential expression using Stringtie and Ballgown.
In the previous module we ran Stringtie in 'reference only' mode using the '-G' and '-e' Stringtie options.
In this module we will run Stringtie in two additional modes: (1) 'reference guided' mode and (2) 'de novo' mode. Stringtie can predict the transcripts present in each library with or without help from knowledge of known transcripts. Stringtie will then assign arbitrary transcript IDs to each transcript assembled from the data and estimate expression for those transcripts. One complication with this method is that in each library, a different set of transcripts is likely to be predicted. There may be a lot of similarities but the number of transcripts and their exact structure will differ in the output files for each library. Before you can compare across libraries you therefore need to determine which transcripts correspond to each other across the libraries. Stringtie provides a merge command to combine predicted transcript GTF files from across different libraries.
Once you have a merged GTF file you can run Stringtie with this instead of the known transcripts GTF file we used before. The merged GTF is used to recalculate expression estimates in prepartion for running Ballgown using the merged, novel transcripts.
To run Stringtie in 'reference guided' mode: use the '-G' option WITHOUT '-e'
To run Stringtie in 'de novo' mode do NOT specify either of the '-G' OR '-e' options.
Refer to the Stringtie manual for a more detailed explanation: https://ccb.jhu.edu/software/stringtie/index.shtml?t=manual
Using the alignments we generated in the previous modules we will now run Stringtie in reference guided mode using the '-G' option ONLY.
Extra options specified below:
- '-p 8' tells Stringtie to use eight CPUs
- '-G ' reference annotation to use for guiding the assembly process (GTF/GFF3)
- '-l' name prefix for output transcripts (default: STRG)
- '-o' output path/file name for the assembled transcripts GTF (default: stdout)
First, create an output directory and then run stringtie in reference-guided mode.
cd $RNA_HOME/
mkdir -p expression/stringtie/ref_guided/
cd expression/stringtie/ref_guided/
stringtie -p 8 -G $RNA_REF_GTF -l HBR_Rep1 -o HBR_Rep1/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep1.bam
stringtie -p 8 -G $RNA_REF_GTF -l HBR_Rep2 -o HBR_Rep2/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep2.bam
stringtie -p 8 -G $RNA_REF_GTF -l HBR_Rep3 -o HBR_Rep3/transcripts.gtf $RNA_ALIGN_DIR/HBR_Rep3.bam
stringtie -p 8 -G $RNA_REF_GTF -l UHR_Rep1 -o UHR_Rep1/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep1.bam
stringtie -p 8 -G $RNA_REF_GTF -l UHR_Rep2 -o UHR_Rep2/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep2.bam
stringtie -p 8 -G $RNA_REF_GTF -l UHR_Rep3 -o UHR_Rep3/transcripts.gtf $RNA_ALIGN_DIR/UHR_Rep3.bam
| Previous Section | This Section | Next Section |
|---|---|---|
| Kallisto | Ref Guided | De novo |