Trinotate Functional Annotation
Now we have a bunch of transcript sequences and have identified some subset of them that appear to be biologically interesting in that they're differentially expressed between our two conditions - but we don't really know what they are or what biological functions they might represent. We can explore their potential functions by functionally annotating them using our Trinotate software and analysis protocol. To learn more about Trinotate, you can visit the Trinotate website.
Again, let's make sure that we're back in our primary working directory called 'trinity_workspace':
% pwd
.
/home/ubuntu/workspace/trinity_workspace
If you're not in the above directory, then relocate yourself to it.
Now, create a Trinotate/ directory and relocate to it. We'll use this as our Trinotate computation workspace.
% mkdir Trinotate
% cd Trinotate
Below, we're going to run a number of different tools to capture information about our transcript sequences.
TransDecoder is a tool we built to identify likely coding regions within transcript sequences. It identifies long open reading frames (ORFs) within transcripts and scores them according to their sequence composition. Those ORFs that encode sequences with compositional properties (codon frequencies) consistent with coding transcripts are reported.
Running TransDecoder is a two-step process. First run the TransDecoder step that identifies all long ORFs.
% $TRANSDECODER_HOME/TransDecoder.LongOrfs -t ../trinity_out_dir/Trinity.fasta
Now, run the step that predicts which ORFs are likely to be coding.
% $TRANSDECODER_HOME/TransDecoder.Predict -t ../trinity_out_dir/Trinity.fasta
You'll now find a number of output files containing 'transdecoder' in their name:
% ls -1 |grep transdecoder
.
Trinity.fasta.transdecoder.bed
Trinity.fasta.transdecoder.cds
Trinity.fasta.transdecoder.gff3
Trinity.fasta.transdecoder.pep
Trinity.fasta.transdecoder_dir/
The file we care about the most here is the 'Trinity.fasta.transdecoder.pep' file, which contains the protein sequences corresponding to the predicted coding regions within the transcripts.
Go ahead and take a look at this file:
% less Trinity.fasta.transdecoder.pep
.
>TRINITY_DN102_c0_g1_i1|m.9 TRINITY_DN102_c0_g1_i1|g.9 ORF TRINITY_DN102_c0_g1_i1|g.9 \
TRINITY_DN102_c0_g1_i1|m.9 type:complete len:185 (+) TRINITY_DN102_c0_g1_i1:23-577(+)
MARYGATSTNPAKSASARGSYLRVSFKNTRETAQAINGWELTKAQKYLDQVLEHQRAIPF
RRYNSSIGRTAQGKEFGVTKARWPAKSVKFIQGLLQNAAANAEAKGLDATKLYVSHIQVN
HAPKQRRRTYRAHGRINKYESSPSHIELVVTEKEEAVAKAAEKKLVRLSSRQRGRIASQK
RITA*
>TRINITY_DN106_c0_g1_i1|m.3 TRINITY_DN106_c0_g1_i1|g.3 ORF TRINITY_DN106_c0_g1_i1|g.3 \
TRINITY_DN106_c0_g1_i1|m.3 type:5prime_partial len:149 (-) TRINITY_DN106_c0_g1_i1:38-484(-)
TNDTNESNTRTMSGNGAQGTKFRISLGLPTGAIMNCADNSGARNLYIMAVKGSGSRLNRL
PAASLGDMVMATVKKGKPELRKKVMPAIVVRQSKAWRRKDGVYLYFEDNAGVIANPKGEM
KGSAITGPVGKECADLWPRVASNSGVVV*
>TRINITY_DN109_c0_g1_i1|m.14 TRINITY_DN109_c0_g1_i1|g.14 ORF TRINITY_DN109_c0_g1_i1|g.14 \
TRINITY_DN109_c0_g1_i1|m.14 type:internal len:102 (+) TRINITY_DN109_c0_g1_i1:2-304(+)
AKVTDLRDAMFAGEHINFTEDRAVYHVALRNRANKPMKVDGVDVAPEVDAVLQHMKEFSE
QVRSGEWKGYTGKKITDVVNIGIGGSDLGPVMVTEALKHYA
>TRINITY_DN113_c0_g1_i1|m.16 TRINITY_DN113_c0_g1_i1|g.16 ORF TRINITY_DN113_c0_g1_i1|g.16 \
TRINITY_DN113_c0_g1_i1|m.16 type:3prime_partial len:123 (-) TRINITY_DN113_c0_g1_i1:2-367(-)
MSDSVTIRTRKVITNPLLARKQFVVDVLHPNRANVSKDELREKLAEAYKAEKDAVSVFGF
RTQFGGGKSTGFGLVYNSVADAKKFEPTYRLVRYGLAEKVEKASRQQRKQKKNRDKKIFG
TG
Type 'q' to exit the 'less' viewer.
There are a few items to take notice of in the above peptide file. The header lines includes the protein identifier composed of the original transcripts along with '|m.(number)'. The 'type' attribute indicates whether the protein is 'complete', containing a start and a stop codon; '5prime_partial', meaning it's missing a start codon and presumably part of the N-terminus; '3prime_partial', meaning it's missing the stop codon and presumably part of the C-terminus; or 'internal', meaning it's both 5prime-partial and 3prime-partial. You'll also see an indicator (+) or (-) to indicate which strand the coding region is found on, along with the coordinates of the ORF in that transcript sequence.
This .pep file will be used for various sequence homology and other bioinformatics analyses below.
Earlier, we ran blastx against our mini SWISSPROT datbase to identify likely full-length transcripts. Let's run blastx again to capture likely homolog information, and we'll lower our E-value threshold to 1e-5 to be less stringent than earlier.
% blastx -db ../data/mini_sprot.pep \
-query ../trinity_out_dir/Trinity.fasta -num_threads 2 \
-max_target_seqs 1 -outfmt 6 -evalue 1e-5 \
> swissprot.blastx.outfmt6
Now, let's look for sequence homologies by just searching our predicted protein sequences rather than using the entire transcript as a target:
% blastp -query Trinity.fasta.transdecoder.pep \
-db ../data/mini_sprot.pep -num_threads 2 \
-max_target_seqs 1 -outfmt 6 -evalue 1e-5 \
> swissprot.blastp.outfmt6
Using our predicted protein sequences, let's also run a HMMER search against the Pfam database, and identify conserved domains that might be indicative or suggestive of function:
% hmmscan --cpu 2 --domtblout TrinotatePFAM.out \
~/CourseData/RNA_data/trinity_trinotate_tutorial/trinotate_data/Pfam-A.hmm \
Trinity.fasta.transdecoder.pep
Note, hmmscan might take a few minutes to run.
The signalP and tmhmm software tools are very useful for predicting signal peptides (secretion signals) and transmembrane domains, respectively.
To predict signal peptides, run signalP like so:
% signalp -f short -n signalp.out Trinity.fasta.transdecoder.pep
Take a look at the output file:
% less signalp.out
.
##gff-version 2
##sequence-name source feature start end score N/A ?
## -----------------------------------------------------------
TRINITY_DN19_c0_g1_i1|m.141 SignalP-4.0 SIGNAL 1 18 0.553 . . YES
TRINITY_DN33_c0_g1_i1|m.174 SignalP-4.0 SIGNAL 1 19 0.631 . . YES
....
How many of your proteins are predicted to encode signal peptides?
Run TMHMM to predict transmembrane regions like so:
% tmhmm --short < Trinity.fasta.transdecoder.pep > tmhmm.out
and examine the output:
% less tmhmm.out
.
TRINITY_DN283_c0_g1::TRINITY_DN283_c0_g1_i1::g.162::m.162 len=308 ExpAA=126.66 First60=11.42 PredHel=6 Topology=o49-71i105-124o134-153i160-182o192-214i227-246o
TRINITY_DN283_c0_g1::TRINITY_DN283_c0_g1_i2::g.163::m.163 len=384 ExpAA=152.47 First60=16.21 PredHel=7 Topology=o44-66i100-119o129-148i155-177o187-209i222-241o344-366i
TRINITY_DN283_c0_g2::TRINITY_DN283_c0_g2_i1::g.164::m.164 len=173 ExpAA=66.94 First60=24.48 PredHel=3 Topology=o35-57i70-92o96-118i
TRINITY_DN284_c0_g2::TRINITY_DN284_c0_g2_i1::g.214::m.214 len=144 ExpAA=0.06 First60=0.04 PredHel=0 Topology=i
TRINITY_DN285_c0_g1::TRINITY_DN285_c0_g1_i1::g.176::m.176 len=100 ExpAA=0.01 First60=0.01 PredHel=0 Topology=o
Generating a Trinotate annotation report involves first loading all of our bioinformatics computational results into a Trinotate SQLite database. The Trinotate software provides a boilerplate SQLite database called 'Trinotate.sqlite' that comes pre-populated with a lot of generic data about SWISSPROT records and Pfam domains (and is a pretty large file consuming several hundred MB). Below, we'll populate this database with all of our bioinformatics computes and our expression data.
As a sanity check, be sure you're currently located in your 'Trinotate/' working directory.
% pwd
.
/home/ubuntu/workspace/trinity_workspace/Trinotate
Copy the provided Trinotate.sqlite boilerplate database into your Trinotate working directory like so:
% cp ~/CourseData/RNA_data/trinity_trinotate_tutorial/trinotate_data/Trinotate.boilerplate.sqlite Trinotate.sqlite
Load your Trinotate.sqlite database with your Trinity transcripts and predicted protein sequences:
% $TRINOTATE_HOME/Trinotate Trinotate.sqlite init \
--gene_trans_map ../trinity_out_dir/Trinity.fasta.gene_trans_map \
--transcript_fasta ../trinity_out_dir/Trinity.fasta \
--transdecoder_pep Trinity.fasta.transdecoder.pep
Load in the various outputs generated earlier:
% $TRINOTATE_HOME/Trinotate Trinotate.sqlite \
LOAD_swissprot_blastx swissprot.blastx.outfmt6
% $TRINOTATE_HOME/Trinotate Trinotate.sqlite \
LOAD_swissprot_blastp swissprot.blastp.outfmt6
% $TRINOTATE_HOME/Trinotate Trinotate.sqlite LOAD_pfam TrinotatePFAM.out
% $TRINOTATE_HOME/Trinotate Trinotate.sqlite LOAD_signalp signalp.out
% $TRINOTATE_HOME/Trinotate Trinotate.sqlite LOAD_tmhmm tmhmm.out
% $TRINOTATE_HOME/Trinotate Trinotate.sqlite report > Trinotate.xls
View the report
% less Trinotate.xls
.
#gene_id transcript_id sprot_Top_BLASTX_hit TrEMBL_Top_BLASTX_hit RNAMMER prot_id prot_coords sprot_Top_BLASTP_hit TrEMBL_Top_BLASTP_hit Pfam SignalP TmHMM eggnog gene_ontology_blast gene_ontology_pfam transcript peptide
TRINITY_DN144_c0_g1 TRINITY_DN144_c0_g1_i1 PUT4_YEAST^PUT4_YEAST^Q:1-198,H:425-490^74.24%ID^E:4e-29^RecName: Full=Proline-specific permease;^Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Saccharomyces . . . .
. . . . . COG0833^permease GO:0016021^cellular_component^integral component of membrane`GO:0005886^cellular_component^plasma membrane`GO:0015193^molecular_function^L-proline transmembrane transporter activity`GO:0015175^molecular_function^neutral amino acid transmembrane transporter activity`GO:0015812^biological_process^gamma-aminobutyric acid transport`GO:0015804^biological_process^neutral amino acid transport`GO:0035524^biological_process^proline transmembrane transport`GO:0015824^biological_process^proline transport . . .
TRINITY_DN179_c0_g1 TRINITY_DN179_c0_g1_i1 ASNS1_YEAST^ASNS1_YEAST^Q:1-168,H:158-213^82.14%ID^E:5e-30^RecName: Full=Asparagine synthetase [glutamine-hydrolyzing] 1;^Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Saccharomyces .
. . . . . . . . COG0367^asparagine synthetase GO:0004066^molecular_function^asparagine synthase (glutamine-hydrolyzing) activity`GO:0005524^molecular_function^ATP binding`GO:0006529^biological_process^asparagine biosynthetic process`GO:0006541^biological_process^glutamine metabolic process`GO:0070981^biological_process^L-asparagine biosynthetic process . . .
TRINITY_DN159_c0_g1 TRINITY_DN159_c0_g1_i1 ENO2_CANGA^ENO2_CANGA^Q:2-523,H:128-301^100%ID^E:4e-126^RecName: Full=Enolase 2;^Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Nakaseomyces; Nakaseomyces/Candida clade . . TRINITY_DN159_c0_g1_i1|m.1 2-523[+] ENO2_CANGA^ENO2_CANGA^Q:1-174,H:128-301^100%ID^E:3e-126^RecName: Full=Enolase 2;^Eukaryota; Fungi; Dikarya; Ascomycota; Saccharomycotina; Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Nakaseomyces; Nakaseomyces/Candida clade . PF00113.17^Enolase_C^Enolase, C-terminal TIM barrel domain^18-174^E:9.2e-79 . . . GO:0005829^cellular_component^cytosol`GO:0000015^cellular_component^phosphopyruvate hydratase complex`GO:0000287^molecular_function^magnesium ion binding`GO:0004634^molecular_function^phosphopyruvate hydratase activity`GO:0006096^biological_process^glycolytic process GO:0000287^molecular_function^magnesium ion binding`GO:0004634^molecular_function^phosphopyruvate hydratase activity`GO:0006096^biological_process^glycolytic process`GO:0000015^cellular_component^phosphopyruvate hydratase complex . .
...
The above file can be very large. It's often useful to load it into a spreadsheet software tools such as MS-Excel. If you have a transcript identifier of interest, you can always just 'grep' to pull out the annotation for that transcript from this report. We'll use TrinotateWeb to interactively explore these data in a web browser below.
Let's use the annotation attributes for the transcripts here as 'names' for the transcripts in the Trinotate database. This will be useful later when using the TrinotateWeb framework.
% $TRINOTATE_HOME/util/annotation_importer/import_transcript_names.pl \
Trinotate.sqlite Trinotate.xls
Nothing exciting to see in running the above command, but know that it's helpful for later on.
Earlier, we generated large sets of tab-delimited files containg lots of data - annotations for transcripts, matrices of expression values, lists of differentially expressed transcripts, etc. We also generated a number of plots in PDF format. These are all useful, but they're not interactive and it's often difficult and cumbersome to extract information of interest during a study. We're developing TrinotateWeb as a web-based interactive system to solve some of these challenges. TrinotateWeb provides heatmaps and various plots of expression data, and includes search functions to quickly access information of interest. Below, we will populate some of the additional information that we need into our Trinotate database, and then run TrinotateWeb and start exploring our data in a web browser.
Once again, verify that you're currently in the Trinotate/ working directory:
% pwd
.
/home/ubuntu/workspace/trinity_workspace/Trinotate
Now, load in the transcript expression data stored in the matrices we built earlier:
% $TRINOTATE_HOME/util/transcript_expression/import_expression_and_DE_results.pl \
--sqlite Trinotate.sqlite \
--transcript_mode \
--samples_file ../samples.txt \
--count_matrix ../Trinity_trans.counts.matrix \
--fpkm_matrix ../Trinity_trans.TMM.EXPR.matrix
Import the DE results from our edgeR_trans/ directory:
% $TRINOTATE_HOME/util/transcript_expression/import_expression_and_DE_results.pl \
--sqlite Trinotate.sqlite \
--transcript_mode \
--samples_file ../samples.txt \
--DE_dir ../edgeR_trans
and Import the clusters of transcripts we extracted earlier based on having similar expression profiles across samples:
% $TRINOTATE_HOME/util/transcript_expression/import_transcript_clusters.pl \
--sqlite Trinotate.sqlite \
--group_name DE_all_vs_all \
--analysis_name diffExpr.P1e-3_C2_clusters_fixed_P_60 \
../edgeR_trans/diffExpr.P1e-3_C2.matrix.RData.clusters_fixed_P_60/*matrix
And now we'll do the same for our gene-level expression and DE results:
% $TRINOTATE_HOME/util/transcript_expression/import_expression_and_DE_results.pl \
--sqlite Trinotate.sqlite \
--gene_mode \
--samples_file ../samples.txt \
--count_matrix ../Trinity_genes.counts.matrix \
--fpkm_matrix ../Trinity_genes.TMM.EXPR.matrix
% $TRINOTATE_HOME/util/transcript_expression/import_expression_and_DE_results.pl \
--sqlite Trinotate.sqlite \
--gene_mode \
--samples_file ../samples.txt \
--DE_dir ../edgeR_gene
Note, in the above gene-loading commands, the term 'component' is used. 'Component' is just another word for 'gene' in the realm of Trinity.
At this point, the Trinotate database should be fully populated and ready to be used by TrinotateWeb.
TrinotateWeb is web-based software and runs locally on the same hardware we've been running all our computes (as opposed to your typical websites that you visit regularly, such as facebook). Launch the mini webserver that drives the TrinotateWeb software like so:
% $TRINOTATE_HOME/run_TrinotateWebserver.pl 3000
Now, visit the following URL in Google Chrome: http://${YOUR_IP_ADDRESS}:3000/cgi-bin/index.cgi
You should see a web form like so:

In the text box, put the path to your Trinotate.sqlite database, as shown above ("/home/ubuntu/workspace/trinity_workspace/Trinotate/Trinotate.sqlite"). Click 'Submit'.
You should now have TrinotateWeb running and serving the content in your Trinotate database:
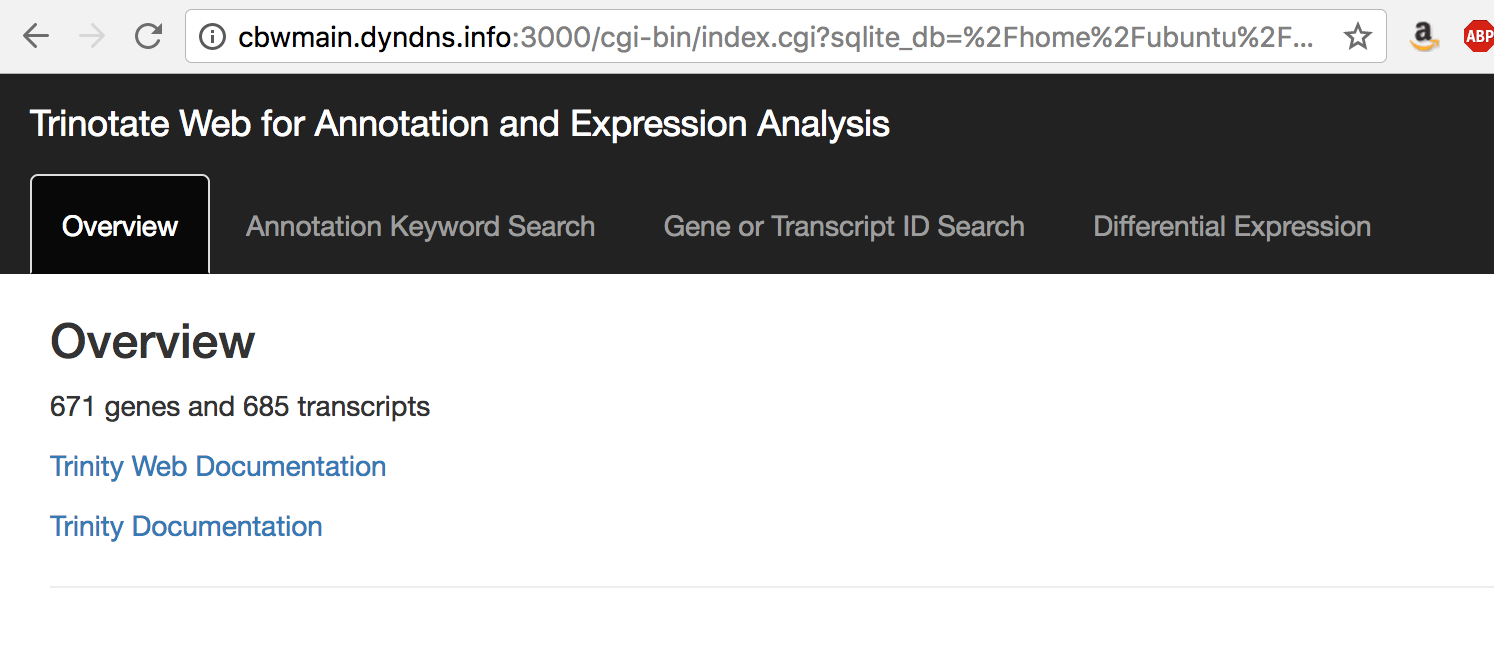
Take some time to click the various tabs and explore what's available.
eg. Under 'Annotation Keyword Search', search for 'transporter'
eg. Under 'Differential Expression', examine your earlier-defined transcript clusters. Also, launch MA or Volcano plots to explore the DE data.
We will explore TrinotateWeb functionality together as a group.
If you've gotten this far, hurray!!! Congratulations!!! You've now experienced the full tour of Trinity and TrinotateWeb. Visit our web documentation at http://trinityrnaseq.github.io, and join our Google group to become part of the ever-growing Trinity user community.
| Previous Section | This Section | Next Section |
|---|---|---|
| Trinity | Trinotate | Saving Results |