This repository contains useful tips/tricks/scripts that I have picked up or developed over the years.
The scripts are mainly written in (bash, awk,perl,R,python), and some of them have been written by by my dear friends and collaborators:
For some of the scripts you have install the following dependencies
sudo apt-get install python3 python3-pip python3-matplotlib \
ipython3-notebook python3-mpltoolkits.basemap
sudo pip3 install -U pip
sudo -H pip3 install --upgrade pandas numpy scipy seaborn
sudo -H pip3 install -U scikit-learn
Author: Augusto César Poot Hernandez,head of the Unidad de Bioinformática y Manejo de la Información of the Instituto de Fisiología Celular, UNAM
Scrip to create a bubble chart from any dataframe contanining either normalized or absolute values.
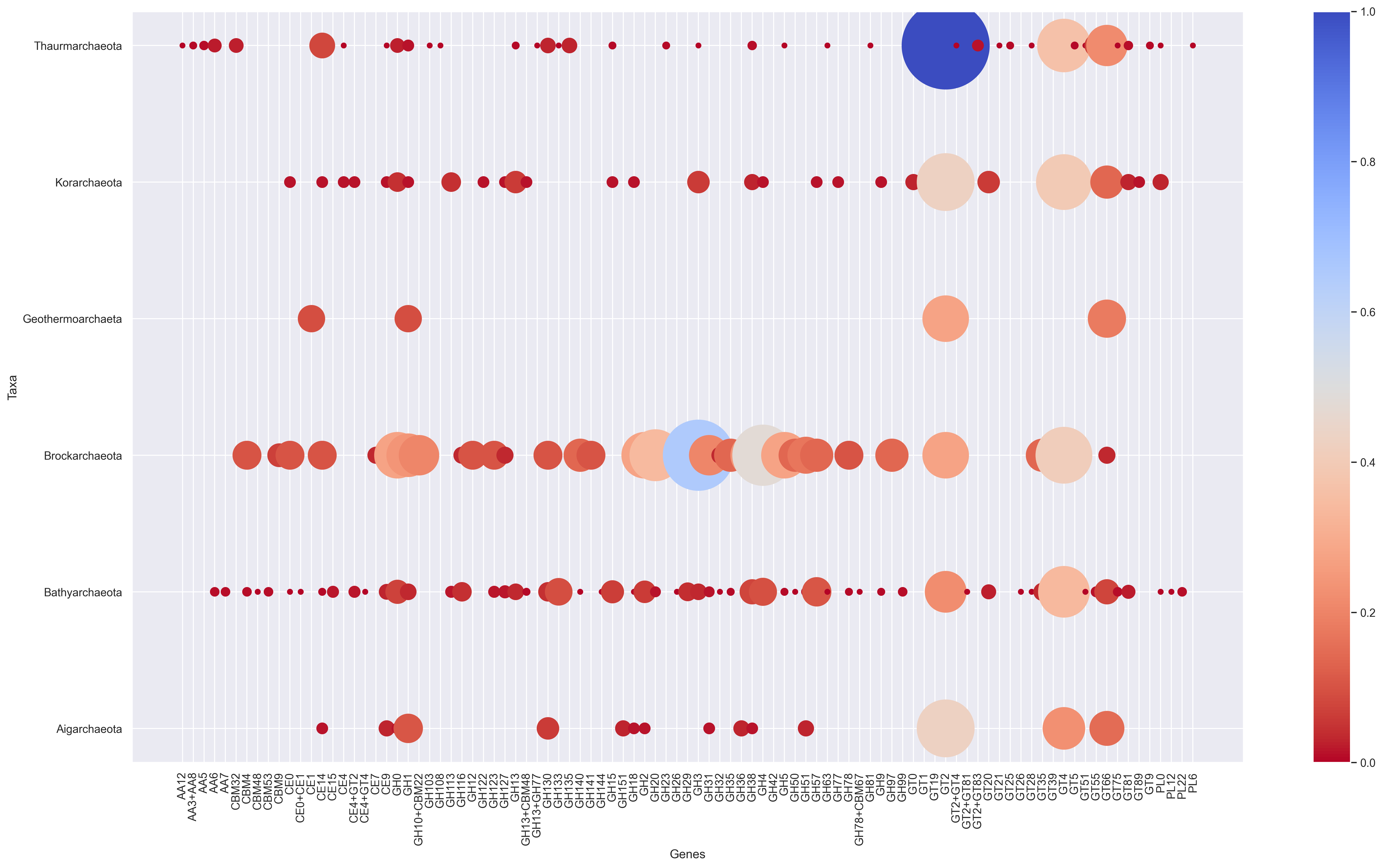
usage: bubble_chart.py [-h] [-im_format {png,pdf,ps,eps,svg,tif,jpg}]
[--im_res dpi]
filename
positional arguments:
filename Input file dataframe i.e abundances profile
optional arguments:
-h, --help show this help message and exit
-im_format {png,pdf,ps,eps,svg,tif,jpg}, -f {png,pdf,ps,eps,svg,tif,jpg}
Output format for images [png].
--im_res dpi, -r dpi Output resolution for images in dot per inch (dpi)
[dpi].Running Bubble plot with example data
python3 bubble_chart.py data_bubbleplot.tab -f png -r 300Customize your script
sns.set(font_scale=1) #change font size
sns.set_style("whitegrid") #whitegrid to change background to white
plt.figure(figsize=(21,12)) #inches, modify to widen (x) or lengthen (y) --> original was 21,12
plt.tight_layout() #keeps axes names in same figure
bubble_super_mega_and_simpe_plot(df, 20, cmap='bone_r', ylabel='Tax Group (# of genomes)', xlabel='Genes',alpha=0.05)
# alpha = transparency
#cmap= color palete see below
#Recomended colors
cmap='bone_r'
cmap='plasma'
cmap='coolwarm_r'
#cmap python = see https://matplotlib.org/3.1.0/tutorials/colors/colormaps.html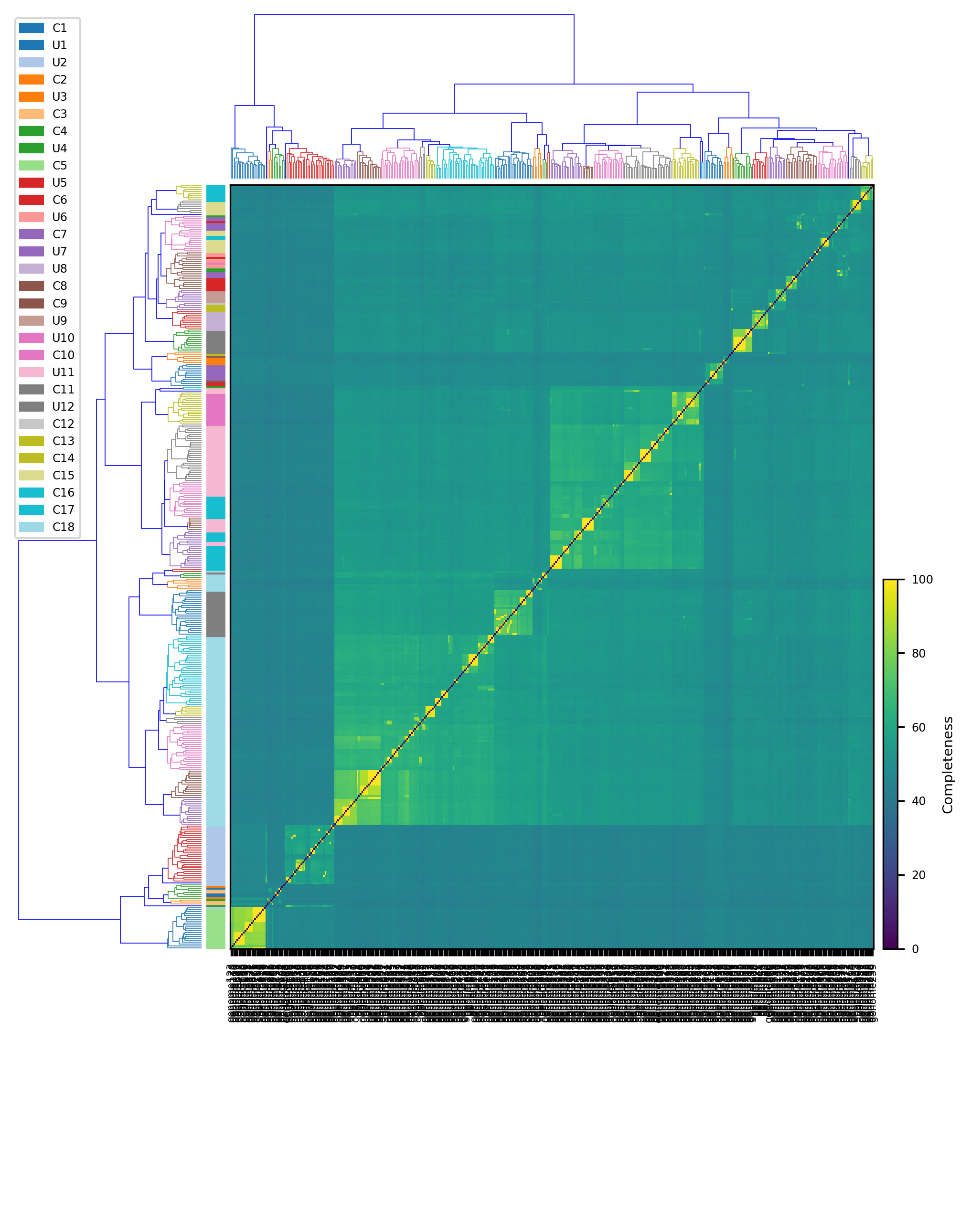
python3 heatmap.py -h
usage: heatmap.py [-h] [-f {png,pdf,ps,eps,svg,tif,jpg}] [-r dpi] filename
This script create a cluster map
positional arguments:
filename Input file derived mebs_output with classification
optional arguments:
-h, --help show this help message and exit
-f {png,pdf,ps,eps,svg,tif,jpg}, --im_format {png,pdf,ps,eps,svg,tif,jpg}
Output format for images [png].
-r dpi, --im_res dpi Output resolution for images in dot per inch (dpi)
[dpi].
Example:
$ python3 heatmap.py data.heatmap.tsvFor Mac OSX users:
Create a conda environment for heatmap.py using the included .yml file heatmap_env_conda_osx.yml:
conda env create -f heatmap_env_conda_osx.yml
A Linux environment will be provided soon.
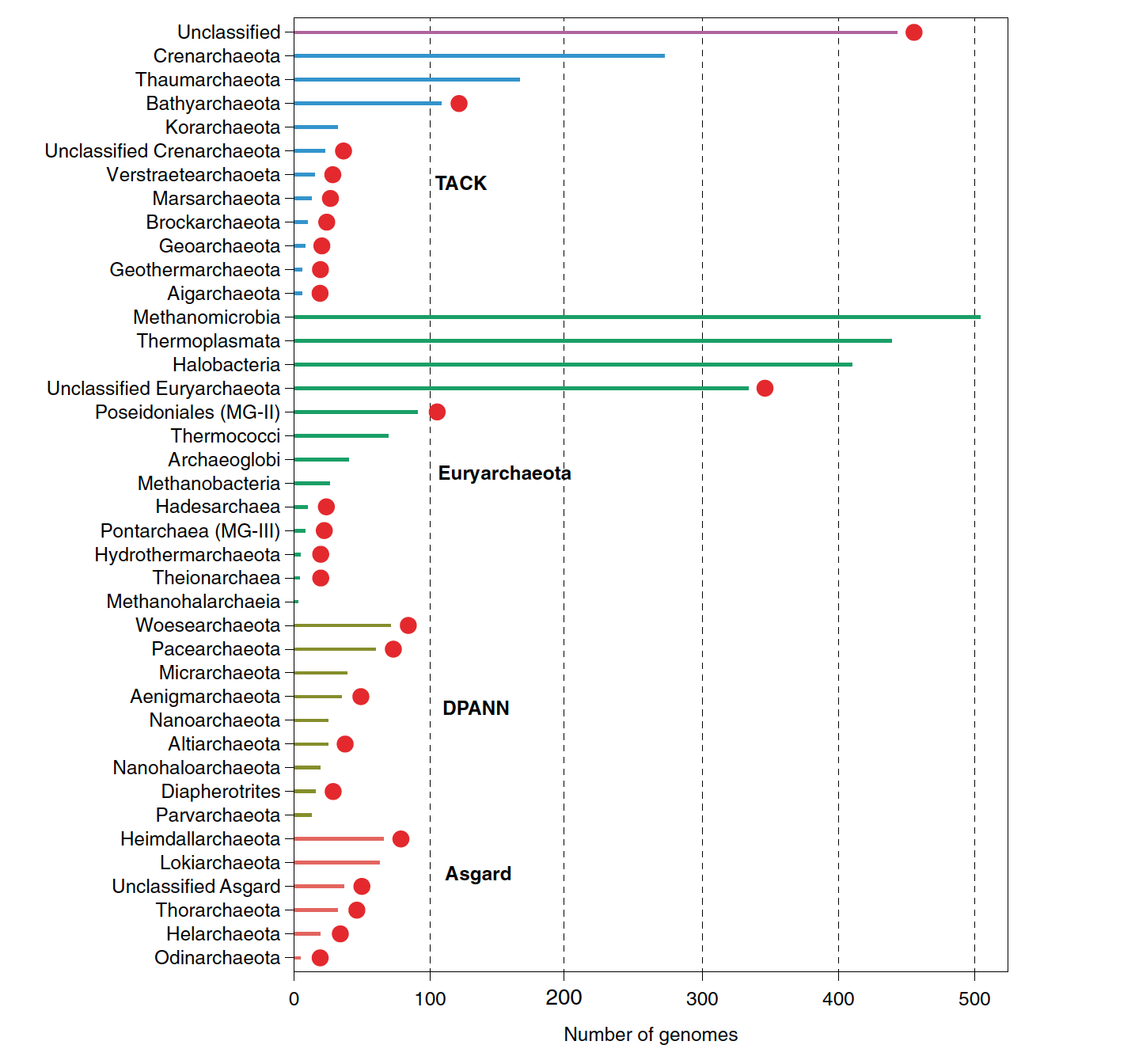
R script that was used to plot the number of achaeal genomes by taxonomy described in Baker et al., 2020
The input data data_barplot.tab, looks like this, the scripts keeps the specific order that you want your data to sorted.
Phylum Superphylum Number of genomes
Heimdallarchaeota Asgard 66
Lokiarchaeota Asgard 63
Unclassified_Asgard Asgard 37
Thorarchaeota Asgard 32
Helarchaeota Asgard 19
barplot.R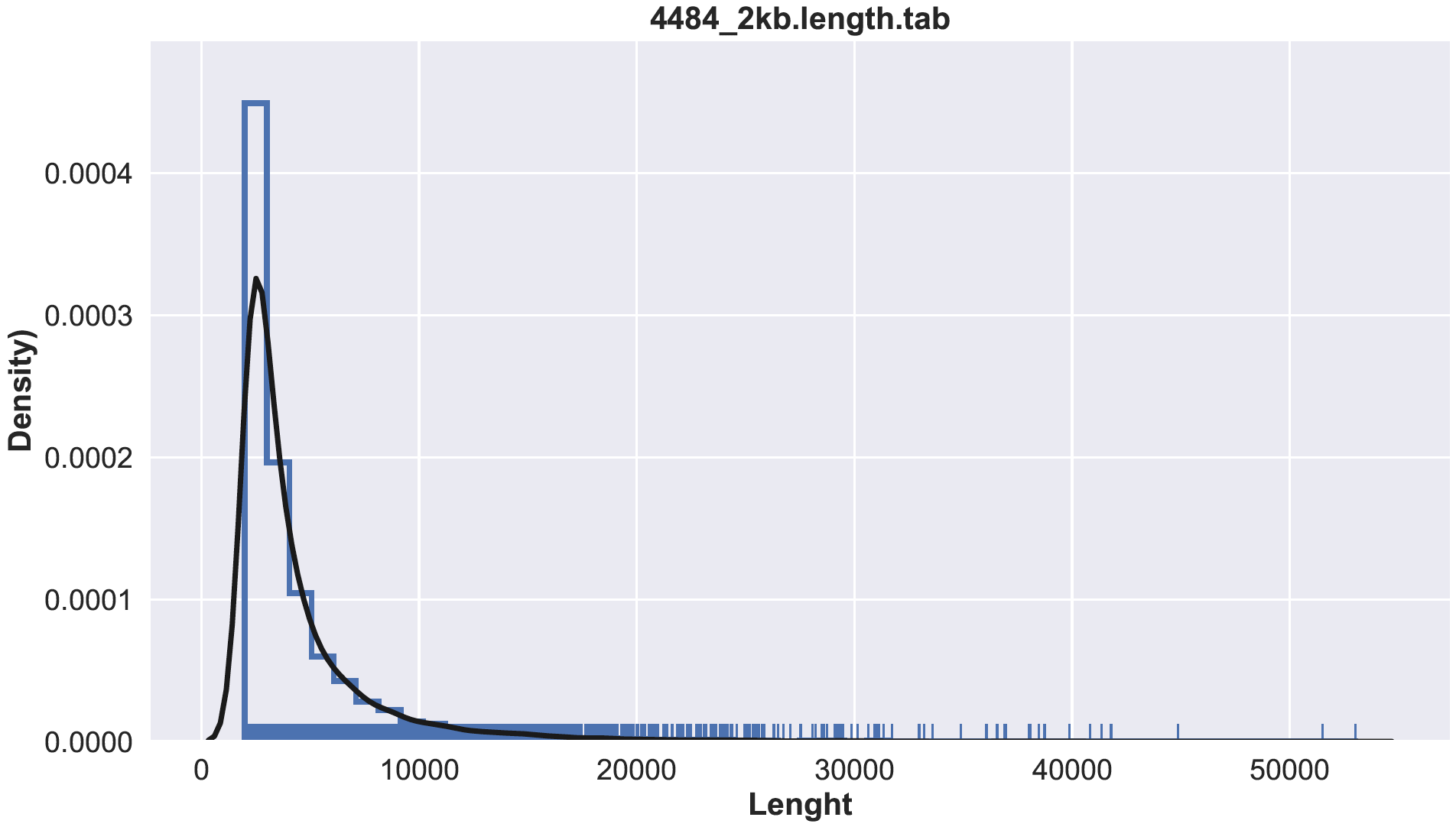
Compute the lenght
seqkit fx2tab --length --name --header-line sample.contigs.fa >> sample.length.tab
less sample.lenght.tab
name length
4484_scaffold_11179 2148
4484_scaffold_8359 2609
4484_scaffold_3616 4460
4484_scaffold_7824 2728
4484_scaffold_6736 3024
4484_scaffold_9058 2482
4484_scaffold_8774 2534
4484_scaffold_4047 4173
4484_scaffold_9826 2344
usage: hist.py [-h] [-im_format {png,pdf,ps,eps,svg,tif,jpg}] [--im_res dpi]
filename
positional arguments:
filename lenght file
optional arguments:
-h, --help show this help message and exit
-im_format {png,pdf,ps,eps,svg,tif,jpg}, -f {png,pdf,ps,eps,svg,tif,jpg}
Output format for images [pdf].
--im_res dpi, -r dpi Output resolution for images in dot per inch (dpi)
[dpi].
Example:
$ python3 histplot.py sample.lenght tab
perl Replace_tree_names.pl mapping_file tree > renamed_treeRequires biopython
pip3 install biopythonScript that is useful if you have a large fasta file and you want to split it into small files of the same size
python3 split_fasta.py
usage: split_fasta.py [-h] [-p PARTS] fastafile
Split a fasta file according in almost equal parts based on total base/residue
count. Stores a numpy array that contains the lengths of the sequences in the
file
positional arguments:
fastafile Fasta file to split
optional arguments:
-h, --help show this help message and exit
-p PARTS, --parts PARTS
Number of parts to slice the file [10]From Tejashree Modak
python3 rename.py --mapping-file MappingFile.csv -i input.fasta -o output.fasta
If you have a directory contanining fasta files (fa: either faa or fna) compute several stats, that are important when describing MAGs See Table 1 Preprint De Anda et al., 2020
for i in *.fa; do seqkit stat $i >> stats; done
for i in *.fa ; do perl gc.pl $i >$i.gc.tab ; done
#Sum the scaffold GC and get the average
for i in *.tab; do awk '{sum+= $2; n++ } END { if (n > 0) print sum / n; }' $i > $i.GC.average ; done
I provide 2 scripts to compute the GC content of fasta sequences. The scripts takes a fasta file as the only parameter
- GC-content.pl
Usage: perl GC-content.pl
The output looks like this
DNA Length is: 590771
Number of G bases: 51234
Number of C bases: 8076
Number of T bases: 24808
Number of A bases: 54731
GC Content is: 10.0394230590195 %
- get_gc_content.pl
Usage: perl get_gc_content.pl
This program takes a fasta file as it's first (and only) parameter.
It returns a tab delimited file (gc_out.txt): column 1 = header ID (everything between ">"
and the first space in the header), and column 2 = gc content for the fasta entry.
Option 1 awk
Obtained from here
cat file.fa | awk '$0 ~ ">" {if (NR > 1) {print c;} c=0;printf substr($0,2,100) "\t"; } $0 !~ ">" {c+=length($0);} END { print c; }' awk '$0 ~ ">" {if (NR > 1) {print c;} c=0;printf substr($0,2,100) "\t"; } $0 !~ ">" {c+=length($0);} END { print c; }' file.faOption 2 Seqkit
seqkit fx2tab --length --name --header-line file.fa >> file.lenght Option3 samtools
samtools faidx file.fa | cut -f1-2 file.fa.fai > file.lenght.tabFrom this
> header 1
ATGCAATGCATG
ATGCCCGGTAGT
TTATAGAGATAG
to this
> header 1
ATGCAATGCATGATGCCCGGTAGTTTATAGAGATAG
perl -lne 'if(/^(>.*)/){ $head=$1 } else { $fa{$head} .= $_ } END{ foreach $s (sort(keys(%fa))){ print "$s\n$fa{$s}\n" }}' file.fa > file1ne.fa perl -lne 'if(/^(>.*)/){$h=$1}else{$fa{$h}.=$_} END{ foreach $h (keys(%fa)){$m+=length($fa{$h})}; printf("%1.0f\t",$m/scalar(keys(%fa))) }' file.faIn this case we are keeping sequences >100 bp
perl -lne 'if(/^(>.*)/){ $head=$1 } else { $fa{$head} .= $_ } END{ foreach $s (keys(%fa)){ print "$s\n$fa{$s}\n" if(length($fa{$s})>100) }}' file.fa > file100.fa perl -lne 'if(/^>(\S+)/){ print ">$ARGV $1"} else{ print }' file.fa > file_renamed.fa
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Let's suppose that you have thousands of genomes and you want to compare the total number of sequences in your genomic dataset. If all your genomes are either .faa or .fna extension, you can use the following one-line command to count the number of sequences and generate a histogram. You can change the figure to pdf, just change pdf("seq.pdf");
grep -c ">" *.faa | sed 's/:/\t/g' | cut -f 2 | Rscript -e 'data=abs(scan(file="stdin")); png("seq.png"); hist(data,xlab="secuences")'It requires a list of headers to remove from a fasta file
List of headers to remove example (sequence_to_remove.txt):
scaffold_142207_c1_687
scaffold_98552_c1_471
scaffold_70258_c1_330
scaffold_155515_c1_771Option 1 python script
python3 remove_sequences.py file.fa sequence_to_remove.txt > file_filtered.fa Option 2 awk
Formating will be different since awk adds spaces, but sequence will be the same as Option 1.
awk 'BEGIN{while((getline<"sequence_to_remove.txt")>0)l[">"$1]=1}/^>/{f=!l[$1]}f' file.fa > file_filtered.faOption 3 grep
Remove only the headers not the entire scaffold if working with metagenomic data.
grep -v -f sequence_to_remove.txt file.fa > file_filtered.fa Option 1 pullseq
pullseq -i file.fa -n sequences_to_extract.txt > extracted_sequences.faOption 2 samtools
cat sequences_to_extract.txt | xargs -n 1 samtools faidx file.fa >> extracted_sequences.fa Option 3 bedtools
Extract fasta with coordinates
sreformat fasta file.fa > file.reformat.fna
bedtools getfasta -fi file.reformat.fna -bed sequences_to_extract_coordinates.tab -fo file_out.faMany options are available here, the one that works for me is this one
while read line
do
if [[ ${line:0:1} == '>' ]]
then
outfile=${line:1:11}.fa
echo $line > $outfile
else
echo $line >> $outfile
fi
done < myseq.fa
For example if you have cleaned a MAG and you want to know which scaffolds were removed use seqkit
- First get the common headers:
seqkit common -s file1.fa file2.fa|grep '>'|cut -c2- > common_ids
- Get all sequences from fasta file1.fa that do not match the IDs in common_ids and store the result in file3.fa:
seqkit grep file1.fa -v -n -f common_ids -o file3.fa
- Explore file3 which has the removed sequences in the clean bin
- Create a list of PFAM identifiers
head identifiers.txt
PF13243
PF13249
PF02458- Run the following commands, originally created by Dr. Carlos Cantalapiedra and incorporated in MEBS
cat identifieres.txt | while read pfam; do
desc=$(curl http://pfam.xfam.org/family/"$pfam"/desc | head -1);
printf "$pfam\t";
printf "$desc\n";
done 2> /dev/null \
> identifiers.desc.tabTake a column of 1 file and another column from another file and create a new file with those columns No need for matching column
paste <(awk '{print $1}' file1.txt ) <(awk '{print $2}' file2.txt ) > file3.txtfor next in $(cat ftp_GCA_download.txt); do wget "$next"; done
or you can do wget -i ftp_GCA_download.txtawk 'x[$1]++ == 1 { print $1 " is duplicated"}'cut -f2,3 file1.txt > file2.txtsed '/pattern/ s/^/replace_pattern/' file.txtsed 's/pattern/replace_pattern/2' file.txtThis is very useful if you have a long header in fasta sequences and you want to get rid of all the characters that aren't useful
sed 's/\s.*$//' file.fa > file2.faIn a file of 2 columns, if 2nd column of file is blank, print 1st column followed by "Your Words", otherwise print 1st and 2nd column, create new file of all this output
awk '{if (!$2) {print $1,"YourWords"} else {print $1, $2}}' > file.tsvGenome browse overview https://www.ncbi.nlm.nih.gov/genome/browse/#!/overview/
Genbank assembly summary file
wget http://ftp.ncbi.nlm.nih.gov/genomes/genbank/assembly_summary_genbank.txtGet the complete and latest genomes from assembly summary genbank
awk -F "\t" '$12=="Complete Genome" && $11=="latest"{print $20}' assembly_summary_genbank.txtModified from Huan Fan's github
- Create an alias in your .bash_profile or .bashrc file with the information of your server
alias server_jupyter='ssh -p XX -L 8000:localhost:8888 [email protected]. XXX'- Once in your server set a secure password to acess your notebooks
jupyter notebook password
- Start jupyter on the remote server
jupyter notebook
- It asks you whether you “Accepting one-time-token-authenticated connection from 127.0.0.1”. I answered ‘__A__laways’ but next time it kept asking me… Then it complains:
Jupyter Notebook requires JavaScript.
Please enable it to proceed.
- Just ingore it buy entering Q. Then your token would be given on the last line, some thing like:
http://localhost:8888/?token=5640c991ffc0c0c6071e9f0d0100d7204e4b05a6d400c440
- Access from your local browser Replace 8888 with 8000, since the later is the port we opened for your local machine, so go to
http://localhost:8000/?token=5640c991ffc0c0c6071e9f0d0100d7204e4b05a6d400c440
on your local browser and you are ready to go!
You can use
or
After searching several options including this package in R, I came across a super friendly to use plattfrom taxonkit - A Cross-platform and Efficient NCBI Taxonomy Toolkit
After installing it, download and uncompress these NCBI taxonoomy file
ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
I downloaded and tar -xvzf the directory in /home/valdeanda/DB/TAXID
Your input file, in this case IDs.txt should look like this
02125
1111708
111780
111781
1147
1284629
165597
1666905
1807358
1827144
1920663
1925591
1933929
To run taxonkit run this
taxonkit lineage --data-dir /home/valdeanda/DB/TAXID/ IDs.txt > IDs.taxonomy.tab
#!/bin/bash
while IFS= read -r line1 <&3 && IFS= read -r line2 <&4;
do ./seqkit common -s OriginalBins/$line1 CleanBins/$line2 |grep '>'| cut -c2- > $line1.common.txt;
done 3<OriginalBins.txt 4<CleanBins.txt
#!/bin/bash
while IFS= read -r line1 <&3 && IFS= read -r line2 <&4
do ./seqkit grep OriginalBins/$line1 -v -n -f $line2 -o $line1.extracted.fa
done 3<OriginalBins.txt 4<OriginalBinsCommon.txt
last revised July 7th 2021
From
https://ncbiinsights.ncbi.nlm.nih.gov/2018/02/22/new-taxonomy-files-available-with-lineage-type-and-host-information/
wget ftp.ncbi.nlm.nih.gov/pub/taxonomy/new_taxdump/new_taxdump.tar.gz
Look at the description of the files in the readme
https://ftp.ncbi.nlm.nih.gov/pub/taxonomy/new_taxdump/taxdump_readme.txt
#1. Install entrez
sudo apt install ncbi-entrez-direct
https://linsalrob.github.io/ComputationalGenomicsManual/Databases/NCBI_Edirect.html
ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/accession2taxid/nucl_gb.accession2taxid.gz
for i in `cat ./acc`; do zgrep -m1 -w "$i" nucl_gb.accession2taxid.gz; done
X68822 X68822.1 9731 1118
Z18640 Z18640.1 9731 1121
Z18643 Z18643.1 27615 1128
Biosample information
for i in `cat Biosample.tab` ; do wget -q -O - "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=biosample&id=$i" > $i ; done
#!/bin/bash
for ACC in `cat Clean_Proteins_acc_number.txt`
do
echo -n -e "$ACC\t"
curl -s "https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=protein&id=${ACC}&rettype=fasta&retmode=xml" |\
grep TSeq_taxid |\
cut -d '>' -f 2 |\
cut -d '<' -f 1 |\
tr -d "\n"
echo
done
Conda is required to use Entrez this way.
#Create an environment in which you can add Entrez Direct
conda create --name entrez
#Activate this new environment
conda activate entrez
#Install Entrez
conda install -c bioconda entrez-direct
#Compile a list of Accession numbers from NCBI (PROTEINS)
less list.txt
ABO08866.1
AFA39020.1
AFA39042.1
AFI78392.1
AOQ24367.1
APC08827.1
ATY72478.1
#Change file to comma separated instead of column
cat list.txt | tr "\n" "," | sed 's/,$//' > list.csv
less list.csv
ABO08866.1,AFA39020.1,AFA39042.1,AFI78392.1,AOQ24367.1,APC08827.1,ATY72478.1
#Make a usable script from the list
sed 's/^/efetch -db protein -format fasta -id /' list.csv > list.sh
less list.sh
efetch -db protein -format fasta -id ABO08866.1,AFA39020.1,AFA39042.1,AFI78392.1,AOQ24367.1,APC08827.1,ATY72478.1
#Run the new script
bash list.sh > list.fahttps://www.metagenomics.wiki/tools/blast/blastn-output-format-6
You will need to use samtools >= 1.9 and bwa >= 0.7.17 If the versions of these tools are outdated on the server, use Conda for a mapping environment.
env_name=mapping_env; conda create -n $env_name && \
conda activate $env_name && \
conda install -c bioconda bwa=0.7.17 samtools=1.9 && \
echo "SUCCESS"You will need the index files in the same location as the fasta assembly file. If not, run following command
#index assembly we want to map to
#combined FASTA containing multiple MAGs in this case
bwa index genomes_combined.fna#see the available parameters
bash bwa_bam_map.sh -h
#assembly we want to map to
#we can map to a combined FASTA containing multiple MAGs in this case
genomes_combined.fna
#reads we want to map
Meg22_1012.fastq.gz
#you can assign an identifier for the run with -s, the output bam and bam index filenames will contain this string
#ensure -b and -k (number of threads for bwa and samtools) do not exceed the number displayed with nproc
bash bwa_bam_map.sh -i genomes_combined.fna -r Meg22_1012.fastq.gz -o read_mapping_output_dir -s genomes_combined-Meg22_1012 -b 40 -k 40 -t /home/profile/tmp -e read_mapping_output_dir/error
This will output coordinate-sorted BAM files, and a BAM index file
You will need to use samtools >= 1.9
bash bam_check.sh -b <directory with BAM files> -o <output directory> -j <number of parallel tests> -n <pattern of BAMs, e.g. Meg22*.bam . Omitting this will analyze all files ending with .bam in the directory>