![]()
NeoLizard is a package for setting up and running a custom pipeline for neoantigen prediction.
The pipeline can start from processed files (vcf/maf/avinput) or from raw sequencing files (fastq/bam).
Both CLI- and web-application versions are available.
Based on intended pipeline usage, install necessary tools.
-FASTQC
-MULTIQC
-CUTADAPT
-ANNOVAR
-MHCFLURRY
-PostgreSQL
- Registration required, download at: https://annovar.openbioinformatics.org/en/latest/user-guide/download
- After downloading, move ANNOVAR into the NeoLizard dir or add to path.
- Assure ANNOVAR works as expected: https://annovar.openbioinformatics.org/en/latest/user-guide/startup
- Make sure ANNOVAR is configured with the desired reference genome, I advise downloading the official hg38.
- perl ./annotate_variation.pl -buildver hg38 -downdb refGene humandb/
- perl ./annotate_variation.pl --buildver hg38 --downdb seq humandb/hg38_seq
- perl ./retrieve_seq_from_fasta.pl humandb/hg38_refGene.txt -seqdir humandb/hg38_seq -format refGene -outfile humandb/hg38_refGeneMrna.fa
- Note: downloads often fail. If necessary, please download all failed downloads manually from https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips.
- Make sure
perlis installed for ANNOVAR usage.
- Install the package:
$ pip install mhcflurry
- If you don't already have it, you will also need to install tensorflow version 2.2.0 or later.
$ pip install tensorflow
or if you're using an Apple silicon based processor (M1,M2,M3):
$ pip install tensorflow-macos
- Fetch the necessary model:
$ mhcflurry-downloads fetch models_class1_presentation
- Note: MHCflurry handles extremely large fasta files poorly (20MB+), be sure to handle them manually for low RAM systems.
- If data collection in a relational database format is intended, please install PostgreSQL.
- A new database will be created by NeoLizard for data storage and classification if needed.
- Install using
pip install
matplotlib==3.6.2
mhcflurry==2.0.6
pandas==1.5.3
plotly==5.15.0
psycopg2==2.9.6
seaborn==0.12.2
setuptools==58.0.4
streamlit==1.24.1
NeoLizard allows a custom pipeline to run in a python environment. At this time, the neoantigen prediction pipeline is fully operational and will create the following directory structure.
input/
├─ file1.maf
├─ file2.maf
output/
├─ avinput_files/
│ ├─ file1.avinput
│ ├─ file2.avinput
├─ NeoLizard.log
├─ predictions.csv
├─ fastas/
│ ├─ file1.fasta
│ ├─ file2.fasta
├─ annotations/
│ ├─ file1.log
│ ├─ file1.variant_function
│ ├─ file2.log
│ ├─ file2.exonic_variant_function
│ ├─ file2.variant_function
│ ├─ file1.exonic_variant_function
- NeoLizard will convert MAF files to .avinput, a custom made input structure that is perfectly compatible with ANNOVAR.
- Using ANNOVAR, NeoLizard will annotate and generate fasta sequences containing the mutations of interest.
- Fasta sequences will be curated and candidate mutation-containing peptide sequences will be generated with corresponding flanks for extra precision.
- Based on MHCflurry trained models, binding affinities, presentation scores and processing scores will be predicted for candidate peptides with corresponding HLA-alleles.
- NeoLizard will gather results in a
.csvfile and store them in a relational format using PostgreSQL if selected.
- Note: if
TCGA_allelesis selected (recommended), NeoLizard will automatically link sample ID's to corresponding HLA-alleles using data from the Pan-Cancer Atlas. - Note: all operations, encountered errors and status updates will be written to 'NeoLizard.log' in the output directory. Make sure to carefully read this upon completion.
The preprocessing pipeline (beta) can be customized using the custom --cmd flag. Make sure the called modules are present in your $PATH. Recommended usage is to perform QC (and adapter removal) using NeoLizard, followed by manual preprocessing based on the generated QC logs. After completion, perform the NeoLizard neoantigen prediction pipeline on resulting vcf/maf files.
Usage: NeoLizard_cli [-h] --input INPUT [--output OUTPUT] [--qc] [--m2a] [--cutadapt] [--cutadapt_commands CUTADAPT_COMMANDS]
[--cutadapt_remove] [--annovar_annotate_variation] [--annovar_coding_change]
[--annovar_coding_change_commands ANNOVAR_CODING_CHANGE_COMMANDS]
[--annovar_annotate_variation_commands ANNOVAR_ANNOTATE_VARIATION_COMMANDS] [--HLA_TCGA]
[--HLA_TCGA_custom HLA_TCGA_CUSTOM] [--HLA_typing] [--mhcflurry] [--add_flanks]
[--peptide_lengths PEPTIDE_LENGTHS [PEPTIDE_LENGTHS ...]] [--TCGA_alleles]
[--custom_alleles CUSTOM_ALLELES [CUSTOM_ALLELES ...]] [--cmd CMD] [--store_db] [--db_username DB_USERNAME]
[--db_password DB_PASSWORD] [--db_host DB_HOST] [--db_name DB_NAME]
optional arguments:
-h, --help show this help message and exit
--input INPUT <Required> Input file(s) path
--output OUTPUT Provide output folder path. If none is specified, current working directory is used.
--qc perform QC
--m2a Convert MAF to AVINPUT
cutadapt:
Cutadapt
--cutadapt Perform cutadapt
--cutadapt_commands CUTADAPT_COMMANDS
Enter commands for cutadapt, excluding input and output, as string e.g. "-q 5 -Q 15,20"
--cutadapt_remove Remove original file(s)
annovar:
Annovar
--annovar_annotate_variation
Perform annotate_variation.
--annovar_coding_change
Perform coding_change.
--annovar_coding_change_commands ANNOVAR_CODING_CHANGE_COMMANDS
Enter commands for annovar, excluding input and output, as string default: "annovar/humandb/hg38_refGene.txt
annovar/humandb/hg38_refGeneMrna.fa --includesnp --onlyAltering --alltranscript --tolerate"
--annovar_annotate_variation_commands ANNOVAR_ANNOTATE_VARIATION_COMMANDS
Enter commands for annovar, excluding input and output, as string default: "-build hg38 -dbtype refGene
annovar/humandb/ --comment"
HLA:
HLA
--HLA_TCGA Use TCGA source for HLA alleles.
--HLA_TCGA_custom HLA_TCGA_CUSTOM
Enter custom path to TCGA HLA alleles text file.
--HLA_typing Perform HLA typing on samples.
mhcflurry:
MHCflurry
--mhcflurry Perform mhcflurry
--add_flanks Generate peptides of given length(s) in sequence and test them --> can add flanks for improved accuracy. (computationally more expensive, only use for low amounts of sequences)
--peptide_lengths PEPTIDE_LENGTHS [PEPTIDE_LENGTHS ...]
Enter length(s) of peptides to scan for, separated by spaces. Default is 9.
--TCGA_alleles Use TCGA PanCancer alleles.
--custom_alleles CUSTOM_ALLELES [CUSTOM_ALLELES ...]
Enter the HLA alleles.
cmd:
Custom command
--cmd CMD Custom command with multiple arguments. Please enter as a string! e.g. 'a_module -m 10 -q 20 -j 4'
database:
Database
--store_db Store results in database. Will be created if not available. Please fill in credentials using
"--db-username" "--db-password" "--db-host" and "--db-name".
--db_username DB_USERNAME
Database username, default superuser is postgres
--db_password DB_PASSWORD
Database password
--db_host DB_HOST Database host, default is localhost
--db_name DB_NAME Database name, lowercase! Default is neolizard_db
Predicting Neoantigens from tumor-normal paired samples using MAF files and TCGA alleles.
python3 neolizard_cli.py --input path_to_maf_files --output output_path --m2a --annovar_annotate_variation --annovar_coding_change --annovar_coding_change_commands --mhcflurry --TCGA_alleles
Predicting Neoantigens from tumor-normal paired samples using MAF files and TCGA alleles, taking into account the flanking sequences for extra precision. (only recommended for low amounts of sequences):
python3 neolizard_cli.py --input path_to_maf_files --output output_path --m2a --annovar_annotate_variation --annovar_coding_change --annovar_coding_change_commands --mhcflurry --TCGA_alleles --add_flanks
Predicting Neoantigens from tumor-normal paired samples using MAF files and TCGA alleles, with custom lengths.
python3 neolizard_cli.py --input path_to_maf_files --output output_path --m2a --annovar_annotate_variation --annovar_coding_change --annovar_coding_change_commands --mhcflurry --TCGA_alleles --peptide_lengths 8 9 10
Predicting Neoantigens from tumor-normal paired samples using MAF files and custom alleles.
python3 neolizard_cli.py --input path_to_maf_files --output output_path --m2a --annovar_annotate_variation --annovar_coding_change --annovar_coding_change_commands --mhcflurry --custom_alleles HLA-A*31:01
Predicting Neoantigens from tumor-normal paired samples using MAF files and TCGA alleles. Storing the results in a PostgreSQL database.
python3 neolizard_cli.py --input path_to_maf_files --output output_path --m2a --annovar_annotate_variation --annovar_coding_change --annovar_coding_change_commands --mhcflurry --TCGA_alleles --store_db --db_username postgres --db_password password123 --db_host localhost --db_name neolizard_db
Performing preprocessing on fastq files (beta):
python3 neolizard_cli.py --input path_to_fastq_files --output output_path --qc --cutadapt --cutadapt_commands '-m 10 -q 20 -j 4' --cutadapt_remove --cmd 'custom_module --some_flag 2 --some_flag 4' --cmd 'custom_module2 --some_other_module --some_flag 2 --some_flag 4'
If preferred, a streamlit python-based browser gui is available for composing the query and visualizing the resulting prediction data. Identical execution as the CLI-based version.
streamlit run neolizard.py

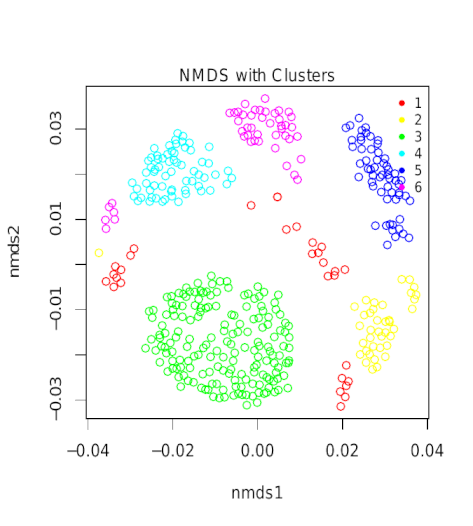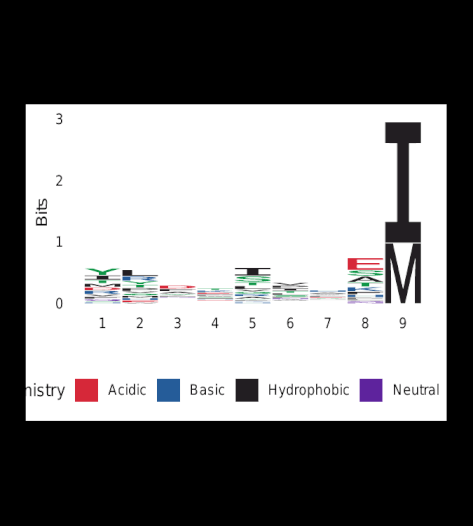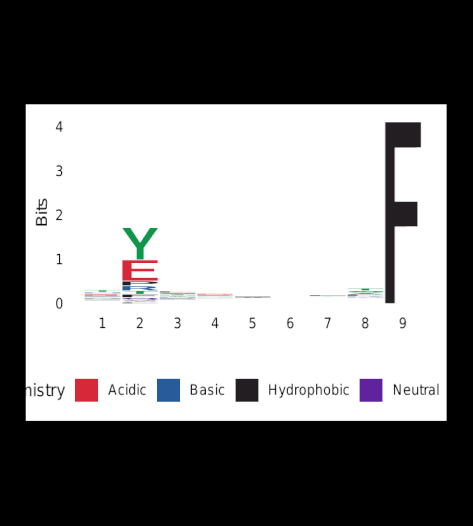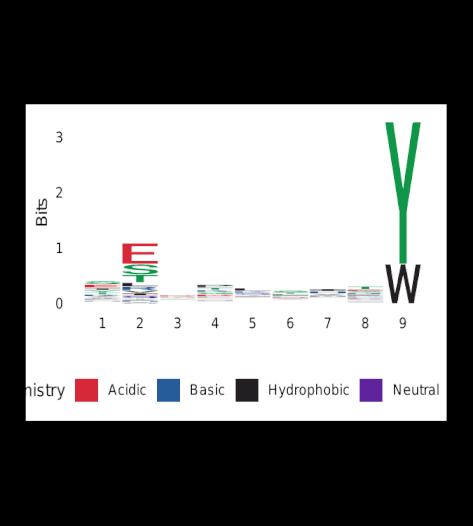
Upon pipeline completion. A range of analyses can be performed on the prediction results. By way of illustration, a pipeline run was performed on the TCGA_PRAD data. This dataset contains about 500 MAF files of Prostate Cancer patients and is open source (https://portal.gdc.cancer.gov/). The MAF files can be downloaded using the gdc-client (https://gdc.cancer.gov/access-data/gdc-data-transfer-tool).
A cluster-analysis was performed on a subset of 500 resulting peptides that showed a binding affinity < 35nM and affinity percentiles < 0.015. These criteria ensure the accuracy of the subset selection.
Upon completion of Gibbs-clustering, sequence-logos were generated for all clusters.
The R-scripts and results can be found respectively in /scripts and /resources/analysis.