-
Notifications
You must be signed in to change notification settings - Fork 1
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
Merge branch 'feature/nextflow_schema' into dev
- Loading branch information
Showing
6 changed files
with
765 additions
and
0 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,3 @@ | ||
| # Changelog | ||
| ## 0.3.0 | ||
| Initial release on CloWM |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,49 @@ | ||
| # Metagenomics-Toolkit | ||
|
|
||
| The Metagenomics-Toolkit is a scalable, data agnostic workflow that automates the analysis of short and long metagenomic reads obtained from Illumina or Oxford Nanopore Technology devices, respectively. | ||
| The Toolkit offers not only standard features expected in a metagenome workflow, such as quality control, assembly, binning, and annotation, but also distinctive features, | ||
| such as plasmid identification based on various tools, the recovery of unassembled microbial community members, and the discovery of microbial interdependencies through a combination of dereplication, co-occurrence, and genome-scale metabolic modeling. | ||
| Furthermore, the Metagenomics-Toolkit includes a machine learning-optimized assembly step that tailors the peak RAM value requested by a metagenome assembler to match actual requirements, thereby minimizing the dependency on dedicated high-memory hardware. | ||
|
|
||
| **Schema of the complete Metagenomics-Toolkit workflow:** | ||
|
|
||
| 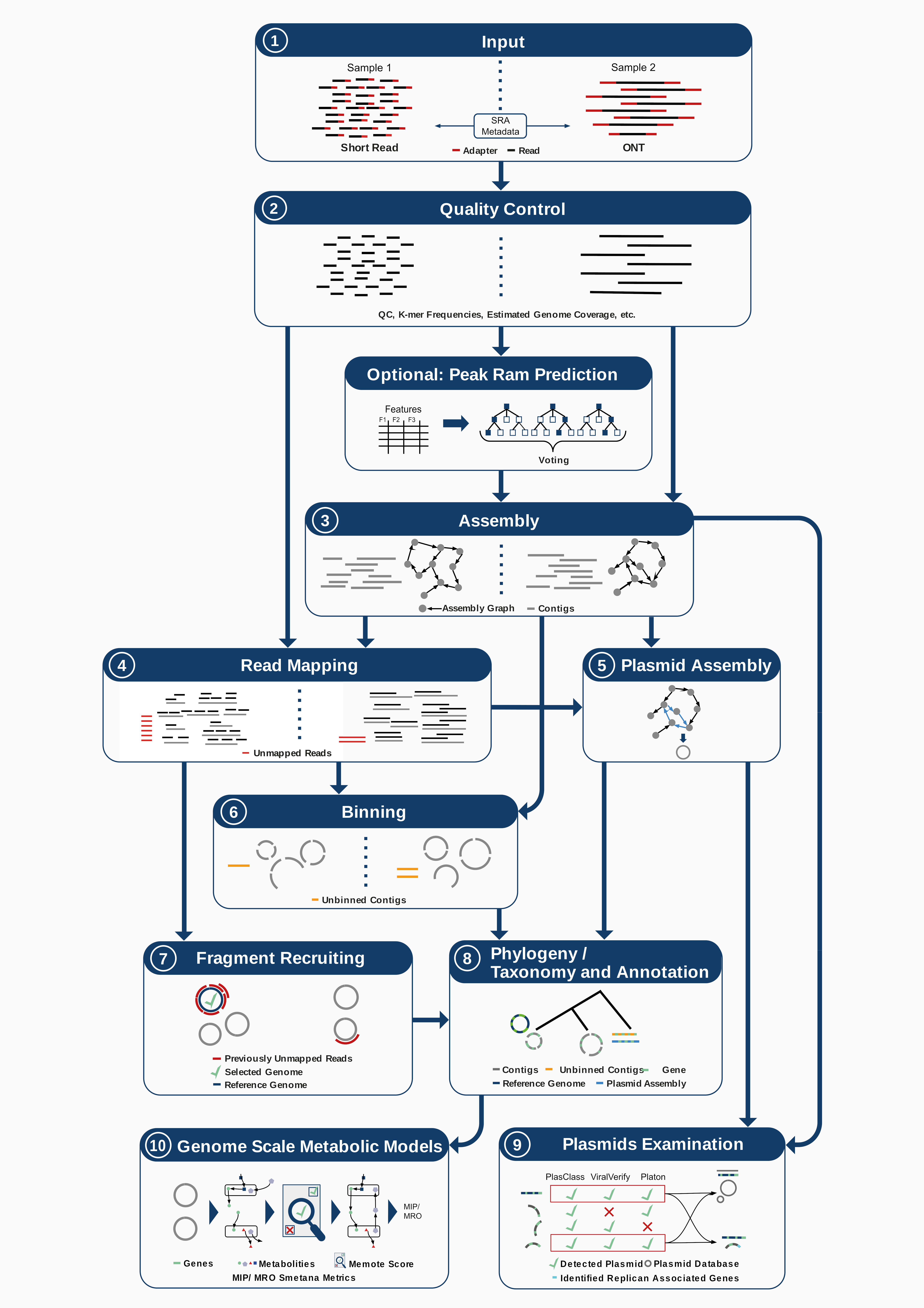 | ||
|
|
||
|
|
||
| > [!IMPORTANT] | ||
| > Below is a list of tools and databases that are enabled for the CloWM service. | ||
| > Currently only the processing of short reads and the corresponding best practice tools are enabled. More tools and databases will be enabled in the future. | ||
| ## Modules and Tools | ||
|
|
||
| Currently the following modules and tools are enabled for execution in CloWM: | ||
|
|
||
| | Box number in figure | Modules | Tools | | ||
| |-----|----|-------| | ||
| | 2 | Quality Control | Fastp, KMC, Nonpareil | | ||
| | 3 | Assembly | MEGAHIT | | ||
| | 4 | Read Mapping | BWA-MEM2 | | ||
| | 6 | Binning | MetaBAT2, MAGScoT | | ||
| | 9 | Plasmids Assembly /Examination | Platon, ViralVerify, PlasClass, PLSDB, SCAPP | | ||
| | 8 | Phylogeny/Taxonomy and Annotation | GTDB-tk, CheckM, Prokka, RGI, MMseqs2, MMSeqs2 taxonomy | | ||
|
|
||
| ### Plasmids | ||
|
|
||
| The plasmid module is able to identify contigs as plasmids and also to assemble plasmids from the sample's FASTQ data. The module is executed in two parts. In the first part contigs of a metagenome assembler are scanned for plasmids. In the second part a plasmid assembler is used to assemble circular plasmids out of raw reads. All plasmid detection tools are executed on the circular assembly result and on the contigs of the metagenome assembler. Only the filtered sequences are used for downstream analysis. | ||
|
|
||
| The identification of plasmids is based on the combined result of tools which have a filter property assigned. The results of all tools with the filter property set to true are combined using either a logical OR or logical AND. | ||
|
|
||
| Example of the OR and AND operations: Let's assume that we have three plasmid detection tools (t1, t2, t3) that have four contigs (c1, c2, c3, c4) as input. Let's further assume that c1 and c2 are detected by all tools as contigs and c3 and c4 are only detected by t1 and t2. By using an AND only c1 and c2 are finally reported by the module as plasmids. By using an OR all contigs would be annotated as plasmids. | ||
|
|
||
| Only the detected plasmids will be used for downstream analysis. | ||
|
|
||
| ## Databases | ||
|
|
||
| The following databases are used: | ||
| - GTDB (Genome Taxonomy Database) | ||
| - VFDB (Virulence Factors Database) | ||
| - KEGG (Kyoto Encyclopedia of Genes and Genomes) | ||
| - bacmet20 (Antibacterial Biocide- and Metal Resistance Genes Database) | ||
| - uniref90 (UniProt Reference Cluster Database) | ||
| - CARD (Comprehensive Antibiotics Resistance Database) | ||
| - PLSD (A plasmid database) |
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,22 @@ | ||
| { | ||
| "inputParameters": ["input.paired.path"], | ||
| "outputParameters": ["output"], | ||
| "resourceParameters": [ | ||
| "steps.magAttributes.gtdb.database.extractedDBPath", | ||
| "steps.magAttributes.checkm.database.extractedDBPath", | ||
| "steps.annotation.mmseqs2.kegg.database.extractedDBPath", | ||
| "steps.annotation.mmseqs2.vfdb.database.extractedDBPath", | ||
| "steps.annotation.mmseqs2.bacmet20_experimental.database.extractedDBPath", | ||
| "steps.annotation.mmseqs2.bacmet20_predicted.database.extractedDBPath", | ||
| "steps.annotation.mmseqs2.uniref90.database.extractedDBPath", | ||
| "steps.annotation.mmseqs2_taxonomy.gtdb.database.extractedDBPath", | ||
| "steps.annotation.rgi.database.extractedDBPath", | ||
| "steps.annotation.keggFromMMseqs2.database.extractedDBPath", | ||
| "steps.plasmid.ViralVerifyPlasmid.database.extractedDBPath", | ||
| "steps.plasmid.PLSDB.database.extractedDBPath", | ||
| "steps.magAttributes.checkm2.database.extractedDBPath" | ||
| ], | ||
| "exampleParameters": { | ||
| "input.paired.path": "https://openstack.cebitec.uni-bielefeld.de:8080/meta_test/test_data/fullPipeline/reads_split.tsv" | ||
| } | ||
| } |
Oops, something went wrong.