{kind=link}
DELVE benchmark is python package designed for evaluating feature selection methods on trajectory preservation in single-cell data.
DELVE is an unsupervised feature selection method for identifying a representative subset of dynamically-expressed molecular features that recapitulate cellular trajectories from single-cell data (e.g. single-cell RNA sequencing, protein iterative immunofluorescence imaging). In contrast to previous work, DELVE uses a bottom-up approach to mitigate the effect of unwanted sources of feature variation confounding inference, and instead models cell states from dynamic feature modules that constitute core regulatory complexes. For more details on the method, please read the associated paper: Ranek JS, Stallaert W, Milner JJ, Redick M, Wolff SC, Beltran AS, Stanley N, and Purvis JE. DELVE: feature selection for preserving biological trajectories in single-cell data. Nature Communications. 2024.
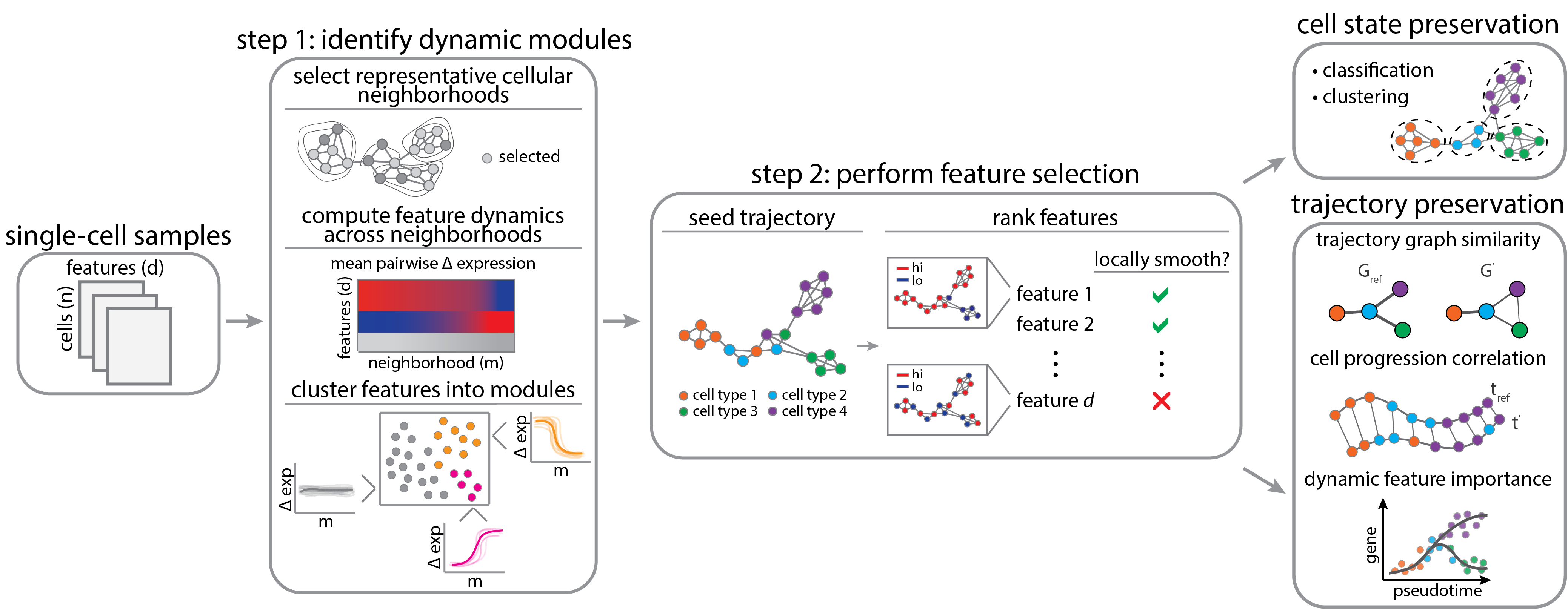
If you'd like to perform feature selection using DELVE, please see the associated repo: https://github.com/jranek/delve. Alternatively, if you'd like to evaluate feature selection methods on trajectory preservation tasks or reproduce the analysis from the paper, please see below.
You can clone the git repository by,
git clone https://github.com/jranek/delve_benchmark.git
Then change the working directory as,
cd delve_benchmark
Given that there are a number of python packages for benchmarking evaluation, we recommend that you create a conda environment using the provided yml file.
conda env create -f venv_delve_benchmark.yml
Once the environment is created, you can activate it by,
conda activate venv_delve_benchmark
If you'd like to perform trajectory inference with Slingshot, or evaluate feature selection methods using simulated data with Splatter or SymSim, you'll need to install the necessary R packages.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
install.packages("devtools")
#slingshot v2.1.1
BiocManager::install("slingshot")
#splatter v1.18.2
BiocManager::install("splatter")
#symsim v0.0.0.9000
library("devtools")
devtools::install_github("YosefLab/SymSim")You can download all of the preprocessed single-cell datasets (.h5ad files) from the Zenodo repository.
We evaluated twelve feature selection methods on their ability to identify features that robustly preserve cell types and cell type transitions from single-cell data. You can compare feature selection methods using the fs class as follows.
This class provides two methods following instantiation, select() which can be used to perform feature selection and evaluate_select() which can be used to evaluate a feature selection method's performance on a trajectory task.
# Parameters
# ----------------------------
# adata: annotated data object containing preprocessed single-cell data (dimensions = cells x features)
# X: array containing preprocessed single-cell data. If None, will access from adata.X
# feature names: list of feature names. If None, will access from adata.var_names
# fs_method: function that specifies feature selection method of interest
# fs_method_params: dictionary that specifies the feature selection method hyperparameters
# eval_method: function that specifies the evaluation method of interest
# eval_method_params: dictionary that specifies the evaluation method hyperparameters
# predicted_features: array containing selected features following feature selection. If None, will perform feature selection according to specified feature selection strategy
# reference_features: array containing reference features for evaluation. If None, will perform random forest classification using provided reference labels
# feature_threshold: number of features to select following feature selection
# feature_threshold_reference: number of features to select from a reference list of features
# labels_key: string referring to the key in adata.obs containing ground truth labels for evaluation
# labels: array containing the ground truth labels for every cell if labels_key is None
# ref_labels: array containing cell type labels if using random forest classification is being used for evaluation
# n_jobs: integer referring to the number of tasks to use in computation
# Method attributes
# ----------------------------
# model.select()
# Performs feature selection according to a specified feature selection method
# Returns:
# predicted features: ranked list of features following feature selection
# model.evaluate_select()
# Performs feature selection according to a feature selection strategy, and then evaluates method performance according to the evaluation criteria or task of interest
# Returns:
# predicted features: ranked list of features following feature selection
# score_df: dataframe of scores
# ----------------------------Here we show you an example of how you can use the fs class to perform feature selection according to a feature selection strategy of interest. We provide 12 functions to perform feature selection. Alternatively, if you'd like to perform feature selection using another method of interest, feel free to add the function to the feature_selection.py script.
delve_benchmark.tl.run_delve_fs- Feature selection for preserving biological trajectories in single-cell datadelve_benchmark.tl.laplacian_score_fs- Laplacian score for feature selectiondelve_benchmark.tl.neighborhood_variance_fs- SLICER: inferring branched, nonlinear cellular trajectories from single cell RNA-seq datadelve_benchmark.tl.mcfs_fs- Unsupervised feature selection for multi-cluster datadelve_benchmark.tl.scmer_fs- Single-cell manifold-preserving feature selection for detecting rare cell populationsdelve_benchmark.tl.hotspot_fs- Hotspot identifies informative gene modules across modalities of single-cell genomicsdelve_benchmark.tl.hvg- Spatial reconstruction of single-cell gene expression datadelve_benchmark.tl.variance_score- Ranks features according to their variancedelve_benchmark.tl.seed_features- Dynamic seed features from Step 1 of the DELVE algorithmdelve_benchmark.tl.all_features- All features without performing selectiondelve_benchmark.tl.random_features- Randomly selected features without replacementdelve_benchmark.tl.random_forest- Ranked feature importance scores from Random Forest classification
To perform feature selection, simply specify the feature selection method and feature selection method hyperparameters. For more details on feature selection method-specific hyperparameters, please see the input parameters in the feature_selection.py script.
import delve_benchmark
import anndata
import os
# Read in preprocessed single-cell data
adata = anndata.read_h5ad(os.path.join('data', 'adata_RPE.h5ad'))
# Example performing feature selection with DELVE
fs = delve_benchmark.tl.fs(adata = adata, fs_method = delve_benchmark.tl.run_delve_fs, fs_method_params = {'num_subsamples': 1000, 'n_clusters': 5, 'k': 10, 'n_random_state': 10})
predicted_features = fs.select()
# Example performing feature selection with the Laplacian Score
fs = delve_benchmark.tl.fs(adata = adata, fs_method = delve_benchmark.tl.laplacian_score_fs, fs_method_params = {'k': 10})
predicted_features = fs.select()We provide 5 functions to evaluate feature selection methods based on their ability to identify features that preserve cell types or cell type transitions. Alternatively, if you'd like to perform evaluation using another evaluation criteria of interest, feel free to add the function to the evaluate.py script. Moreover, please see the evaluate.py script if you'd like to see additional evaluation criteria used in the paper.
delve_benchmark.tl.pak- computes precision @ k score between selected features and ground truth reference featuresdelve_benchmark.tl.svm- performs support vector machine classification using selected features, and then computes the accuracy between predicted and ground truth cell type labelsdelve_benchmark.tl.svm_svr- performs support vector machine regression using selected features, and then computes the mean squared error between predicted and ground truth cell progressiondelve_benchmark.tl.kt_corr- computes the Kendall rank correlation between pseudotime estimated following feature selection and ground truth progressiondelve_benchmark.tl.cluster- performs KMeans clustering on selected features, and then computes NMI score between predicted and ground truth cell type labels
To perform evaluation, simply specify the feature selection method, feature selection method hyperparameters, evaluation method, and the evaluation method hyperparameters. For more details on evaluation method-specific hyperparameters, please see the input parameters in the evaluate.py script.
# Example evaluating DELVE feature selection performance on preserving cell types using SVM classification
fs = delve_benchmark.tl.fs(adata = adata, fs_method = delve_benchmark.tl.run_delve_fs, fs_method_params = {'num_subsamples': 1000, 'n_clusters': 5, 'k': 10, 'n_random_state': 10},
eval_method = delve_benchmark.tl.svm, eval_method_params = {'n_splits': 10}, labels_key = 'phase', feature_threshold = 30)
predicted_features, scores = fs.evaluate_select()
# Example evaluating hotspot feature selection performance on clustering
fs = delve_benchmark.tl.fs(adata = adata, fs_method = delve_benchmark.tl.hotspot_fs, fs_method_params = {'k': 10, 'n_pcs': 50, 'model': 'danb'},
eval_method = delve_benchmark.tl.cluster, eval_method_params = {'n_clusters': 4, 'n_sweep': 25}, labels_key = 'phase', feature_threshold = 30)
predicted_features, scores = fs.evaluate_select()This software is licensed under the MIT license (https://opensource.org/licenses/MIT).