Java utilities for Next Generation Sequencing
Pierre Lindenbaum PhD
http://plindenbaum.blogspot.com
@yokofakun
Tested with java 1.7 and the Picard library 1.91 ( http://sourceforge.net/projects/picard/)
git clone "https://github.com/lindenb/jvarkit.git"
cd jvarkit.gitedit build.properties to configure the project. Something like:
picard.version=1.91
picard.dir=/home/lindenb/package/picard-tools-${picard.version}
picard.jar=${picard.dir}/picard-${picard.version}.jar
sam.jar=${picard.dir}/sam-${picard.version}.jar
variant.jar=${picard.dir}/variant-${picard.version}.jar
tribble.jar=${picard.dir}/tribble-${picard.version}.jar
Working behind a proxy ? Edit the file build.dtd. Change it to:
<!ENTITY httpProxyHost "proxy-upgrade.univ-nantes.prive">
<!ENTITY httpProxyPort "3128">
<!ENTITY xjcproxyarg " <arg value='-httpproxy'/> <arg value='&httpProxyHost;:&httpProxyPort;'/>">
(...)no proxy, set build.dtd to
<!ENTITY httpProxyHost "">
<!ENTITY httpProxyPort "">
<!ENTITY xjcproxyarg "">
(...)| Option | Description |
|---|---|
| SCRIPT_FILE=File | javascript file Default value: null. |
| SCRIPT_EXPRESSION=String | javascript expression Default value: null. |
| IN=String | VCF file/URL to process. Default stdin. Default value: null. |
| OUT=File | VCF file to generate. Default stdout. Default value: null. |
the script binds the following variables:
- variant : the current variation; a org.broadinstitute.variant.variantcontext.VariantContext ( http://sourceforge.net/p/picard/code/HEAD/tree/trunk/src/java/org/broadinstitute/variant/variantcontext/VariantContext.java )
- header : the VCF header org.broadinstitute.variant.vcf.VCFHeader ( http://sourceforge.net/p/picard/code/HEAD/tree/trunk/src/java/org/broadinstitute/variant/vcf/VCFHeader.java).
the script should return '1' or true if the current VCF file should be printed.
the file filter.js
/** prints a VARIATION if two samples at least
have a DP<200 */
function myfilterFunction()
{
var samples=header.genotypeSamples;
var countOkDp=0;
for(var i=0; i< samples.size();++i)
{
var sampleName=samples.get(i);
if(! variant.hasGenotype(sampleName)) continue;
var genotype = variant.genotypes.get(sampleName);
if( ! genotype.hasDP()) continue;
var dp= genotype.getDP();
if(dp < 200 ) countOkDp++;
}
return (countOkDp>2)
}
myfilterFunction();$ curl -s "https://raw.github.com/jamescasbon/PyVCF/master/vcf/test/gatk.vcf" |\
java -jar dist/vcffilterjs.jar SCRIPT_FILE=filter.js
##fileformat=VCFv4.1
##FORMAT=<ID=AD,Number=.,Type=Integer,Description="Allelic depths for the ref and alt alleles in the order listed">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth (reads with MQ=255 or with bad mates are filtered)">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT BLANK NA12878 NA12891 NA12892 NA19238 NA19239 NA19240
chr22 42526449 . T A 151.47 . AC=1;AF=0.071;AN=14;BaseQRankSum=2.662;DP=1226;DS;Dels=0.00;FS=0.000;HRun=0;HaplotypeScore=41.2083;MQ=240.47;MQ0=0;MQRankSum=0.578;QD=4.89;ReadPosRankSum=3.611 GT:AD:DP:GQ:PL 0/1:23,8:31:99:190,0,694 0/0:188,0:190:99:0,478,5376 0/0:187,0:187:99:0,493,5322 0/0:247,0:249:99:0,634,6728 0/0:185,0:185:99:0,487,5515 0/0:202,0:202:99:0,520,5857 0/0:181,1:182:99:0,440,5362
chr22 42526634 . T C 32.60 . AC=1;AF=0.071;AN=14;BaseQRankSum=1.147;DP=1225;DS;Dels=0.00;FS=0.000;HRun=0;HaplotypeScore=50.0151;MQ=240.65;MQ0=0;MQRankSum=1.151;QD=1.30;ReadPosRankSum=1.276 GT:AD:DP:GQ:PL 0/1:21,4:25:71:71,0,702 0/0:187,2:189:99:0,481,6080 0/0:233,0:233:99:0,667,7351 0/0:230,0:230:99:0,667,7394 0/0:174,1:175:99:0,446,5469 0/0:194,2:196:99:0,498,6239 0/0:174,0:175:99:0,511,5894
chr22 42527793 rs1080989 C T 3454.66 . AC=2;AF=0.167;AN=12;BaseQRankSum=-3.007;DB;DP=1074;DS;Dels=0.01;FS=0.000;HRun=1;HaplotypeScore=75.7865;MQ=209.00;MQ0=0;MQRankSum=3.014;QD=9.36;ReadPosRankSum=0.618 GT:AD:DP:GQ:PL ./. 0/1:72,90:162:99:1699,0,1767 0/1:103,96:202:99:1756,0,2532 0/0:188,0:188:99:0,526,5889 0/0:160,0:160:99:0,457,4983 0/0:197,0:198:99:0,544,6100 0/0:156,0:156:99:0,439,5041
$ ant vcffilterjs
Buildfile: /home/lindenb/src/jvarkit-git/build.xml
vcffilterjs:
(...)
BUILD SUCCESSFUL
Total time: 1 second
"Sequence logo ( http://weblogo.berkeley.edu/logo.cgi ) for different alleles or generated from SAM/BAM" http://www.biostars.org/p/73021
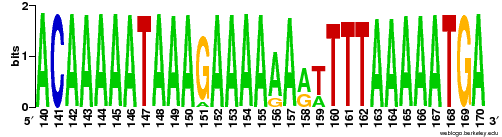
ant sam4weblogo- INPUT=File A BAM file to process. Required.
- REGION=String Region to observe: chrom:start-end Required.
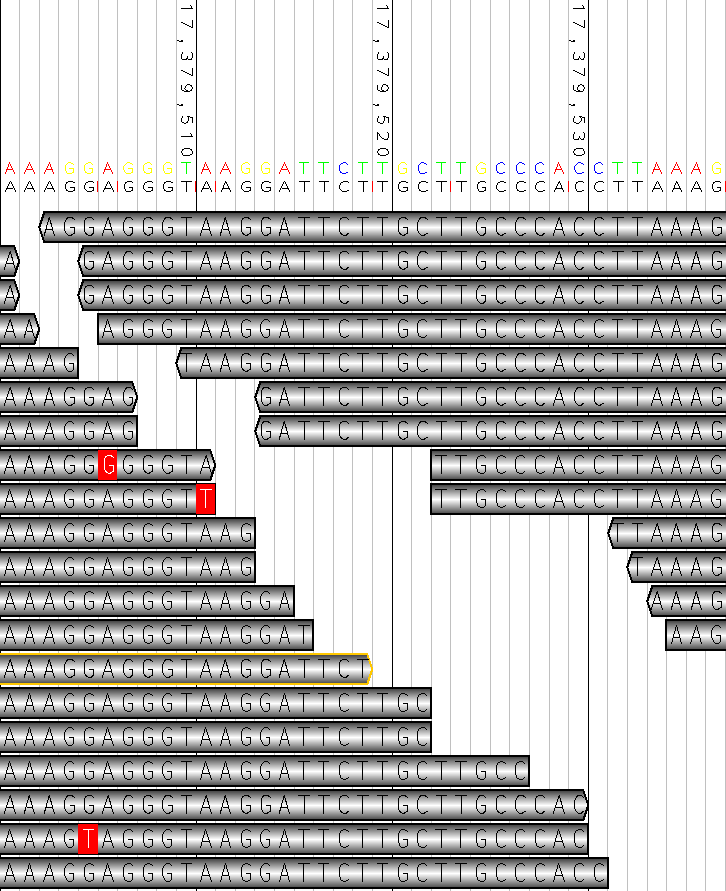
display a tabular view of each base of the reads vs the reference. ```bash ant sam2tsv ```
- IN=File BAM files to process. This option may be specified 0 or more times.
- REGION=String restrict to that region (chr:start-end) Default value: null.
- REF=File Indexed reference Required.
- A={true,false} Use Alignment format.
M00491:12:000000000-A3FL3:1:1101:16929:4287 147 1 A 20 chr22 544289 A M = M00491:12:000000000-A3FL3:1:1101:16929:4287 147 2 G 28 chr22 544290 G M = M00491:12:000000000-A3FL3:1:1101:16929:4287 147 3 A 32 chr22 544291 C M X M00491:12:000000000-A3FL3:1:1101:16929:4287 147 4 T 37 chr22 544292 T M = M00491:12:000000000-A3FL3:1:1101:16929:4287 147 5 C 36 chr22 544293 C M =
<br/>
<h3>cmpbam: Comparing two or more BAMS</h3>
<h4>Compilation</h4>
```bash
ant cmpbams
| Option | Description |
|---|---|
| IN=File | BAM files to process. This option must be specified at least 2 times. |
| REGION=String | restrict to that region (chr:start-end) Default value: null. |
| USESAMFLAG=Boolean | use SAM Flag when comparing. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
(under development) Save a BAM alignment as a PNG image.
| Option | Description |
|---|---|
| IN=File | BAM files to process. Required. |
| OUT=File | Image name. Required. |
| REGION=String | restrict to that region (chr:start-end) Default value: null. |
| REF=File | Indexex reference Required. |
| WIDTH=Integer | image width Default value: 1000. This option can be set to 'null' to clear the default value. |
| NAME=Boolean | print read name. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| BASE=Boolean | print base. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
{kind=link}
Generate the SQL code to insert a VCF into sqlite3.
| Option | Description |
|---|---|
| IN=File | VCF files to process. This option may be specified 0 or more times. |
| SUFFIX=String | Table suffix Default value: (empty). |
| USE_VEP=Boolean | Use and explode VEP predictions Default value: true. Possible values: {true, false} |
| USE_SNPEFF=Boolean | Use and explode SNPEFF predictions Default value: true. Possible values: {true, false} |
| ENGINE=String | sql engine [sqlite,hsql] Default value: sqlite. This option can be set to 'null' to clear the default value. |
| SPLIT4=Boolean | Split DP4 Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| Option | Description |
|---|---|
| IN=File | VCF file (or stdin). Default value: null. |
| OUT=File | output file (or stdout). Default value: null. |
| REF=File | Reference file. Required. |
Filters a BAM using javascript( java rhino engine). The script puts 'record' a SamRecord (http://picard.sourceforge.net/javadoc/net/sf/samtools/SAMRecord.html) and 'header' ( http://picard.sourceforge.net/javadoc/net/sf/samtools/SAMFileHeader.html) in the script context
| Option | Description |
|---|---|
| IN=File | BAM file to process. Default stdin. |
| OUT=File | output filename. Default stdin. |
| SCRIPT_FILE=File | javascript file |
| SCRIPT_EXPRESSION=String | javascript expression |
| SAM_OUTPUT=Boolean | sam output Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| LIMIT=Long | limit to 'L' records. |
java -jar dist/samjs.jar I=ex1.bam \
VALIDATION_STRINGENCY=SILENT \
SCRIPT_EXPRESSION="record.readUnmappedFlag || record.mateUnmappedFlag;" \
SAM=true
@HD VN:1.4 SO:unsorted
@SQ SN:seq1 LN:1575
@SQ SN:seq2 LN:1584
B7_591:4:96:693:509 73 seq1 1 99 36M * 0 0 CACTAGTGGCTCATTGTAAATGTGTGGTTTAACTCG <<<<<<<<<<<<<<<;<<<<<<<<<5<<<<<;:<;7 H0:i:1 H1:i:0 MF:i:18 NM:i:0 UQ:i:0 Aq:i:73
EAS54_65:7:152:368:113 73 seq1 3 99 35M * 0 0 CTAGTGGCTCATTGTAAATGTGTGGTTTAACTCGT <<<<<<<<<<0<<<<655<<7<<<:9<<3/:<6): H0:i:1 H1:i:0 MF:i:18 NM:i:0 UQ:i:0 Aq:i:66
EAS51_64:8:5:734:57 137 seq1 5 99 35M * 0 0 AGTGGCTCATTGTAAATGTGTGGTTTAACTCGTCC <<<<<<<<<<<7;71<<;<;;<7;<<3;);3*8/5 H0:i:1 H1:i:0 MF:i:18 NM:i:0 UQ:i:0 Aq:i:66
B7_591:1:289:587:906 137 seq1 6 63 36M * 0 0 GTGGCTCATTGTAATTTTTTGTTTTAACTCTTCTCT (-&----,----)-)-),'--)---',+-,),''*, H0:i:0 H1:i:0 MF:i:130 NM:i:5 UQ:i:38 Aq:i:63
EAS56_59:8:38:671:758 137 seq1 9 99 35M * 0 0 GCTCATTGTAAATGTGTGGTTTAACTCGTCCATGG <<<<<<<<<<<<<<<;<;7<<<<<<<<7<<;:<5% H0:i:1 H1:i:0 MF:i:18 NM:i:0 UQ:i:0 Aq:i:72
EAS56_61:6:18:467:281 73 seq1 13 99 35M * 0 0 ATTGTAAATGTGTGGTTTAACTCGTCCCTGGCCCA <<<<<<<<;<<<8<<<<<;8:;6/686&;(16666 H0:i:0 H1:i:1 MF:i:18 NM:i:1 UQ:i:5 Aq:i:39
EAS114_28:5:296:340:699 137 seq1 13 99 36M * 0 0 ATTGTAAATGTGTGGTTTAACTCGTCCATGGCCCAG <<<<<;<<<;<;<<<<<<<<<<<8<8<3<8;<;<0; H0:i:1 H1:i:0 MF:i:18 NM:i:0 UQ:i:0 Aq:i:73
B7_597:6:194:894:408 73 seq1 15 99 35M * 0 0 TGTAAATGTGTGGTTTAACTCGTCCATTGCCCAGC <<<<<<<<<7<<;<<<<;<<<7;;<<<*,;;572< H0:i:0 H1:i:1 MF:i:18 NM:i:1 UQ:i:9 Aq:i:43
EAS188_4:8:12:628:973 89 seq1 18 75 35M * 0 0 AAATGTGTGGTTTAACTCGTCCATGGCCCAGCATT ==;=:;:;;:====;=;===:=======;==;=== H0:i:1 H1:i:0 MF:i:64 NM:i:0 UQ:i:0 Aq:i:0
(...)creates a table for DESEQ with the number of reads within a sliding window for multiple BAMS.Version: 1.0 ```bash ant bam4deseq01 ```
| Option | Description |
|---|---|
| IN=File | BAM file to process This option must be specified at least 1 times. |
| OUT=File | output filename. Default stdout. |
| WINDOW_SIZE=Integer | size of the observed window. Default value: 500. This option can be set to 'null' to clear the default value. |
| WINDOW_SHIFT=Integer | shift window by SHIFT pb Default value: 250. This option can be set to 'null' to clear the default value. |
| ONLY_COVERED=Boolean | ignore regions with NO coverage Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| HEADER=Boolean | print header Default value: true. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
Removes one or more field from the INFO column of a VCF.Version ```bash ant vcfstripannot ```
| Option | Description |
|---|---|
| IN=File | VCF file to process. Default stdin. |
| OUT=File | VCF file to generate. Default stdout. Default value: null. |
| KEY=String | remove this INFO key This option may be specified 0 or more times. |
| RESET_FILTER=Boolean | Reset the FILTER column Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
```bash $ java -jar dist/vcfstripannot.jar K=DP4 K=AC1 < in.vcf > out.vcf ``` Fix samtools INDELS for @SolenaLS ```bash ant vcffixindels ```
| Option | Description |
|---|---|
| IN=File | VCF file to process. Default stdin. Default value: null. |
| OUT=File | VCF file to generate. Default stdout. Default value: null. |
```bash $ java -jar dist/vcffixindels.jar I=input.vcf.gz | grep FIX
2 749862 . T TT 94.50 . AC1=2;AF1=1;DP=13;DP4=0,0,5,8;FQ=-73.5;INDEL;INDELFIXED=CTTTTTT|CTTTTTTT|74954856;IS=13,1.000000;MQ=50;VDB=4.019414e-04 GT:PL:DP:GQ 1/1:135,39,0:13:75
<h3>BlastMapAnnots</h3>
<h4>Motivation</h4>
Maps uniprot/genbank annotations on a blast result. See http://www.biostars.org/p/76056
<h4>Compilation</h4>
This tools call ${JAVA_HOME}/bin/xjc. If you're working being a proxy, you might have to edit the build.xml file to add the -httpproxy option.
```bash
ant blastmapannots
| Option | Description |
|---|---|
| IN=File | XML sequence file Genbank.xml or uniprot.xml. Required. |
| BLAST=File | BLAST XML output (or stdin). Default value: null. |
| APPEND_ACN=Boolean | append the sequence accession before the feature name. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| INCL=String | Restrict to uniprot/feature/type of genbank/feature/key. This option may be specified 0 or more times. |
| EXCL=String | Exclude uniprot/feature/type of genbank/feature/key. This option may be specified 0 or more times. |
Download uniprot P04514 ( Rotavirus Non-structural protein 3 ) as XML ```bash $ curl -o P04514.xml "http://www.uniprot.org/uniprot/P04514.xml" ``` Download the same P04514 as fasta ```bash $ curl -o P04514.fasta "http://www.uniprot.org/uniprot/P04514.fasta" ```
TblastN P04514.fasta vs a RNA of NSP3 in genbank http://www.ncbi.nlm.nih.gov/nuccore/AY065842.1 and save the ouput as XML:
<?xml version="1.0"?>
<!DOCTYPE BlastOutput PUBLIC "-//NCBI//NCBI BlastOutput/EN" "http://www.ncbi.nlm.nih.gov/dtd/NCBI_BlastOutput.dtd">
<BlastOutput>
<BlastOutput_program>tblastn</BlastOutput_program>
(...)
<Hit>
<Hit_num>1</Hit_num>
<Hit_id>gi|18139606|gb|AY065842.1|</Hit_id>
<Hit_def>Rhesus rotavirus nonstructural protein 3 (NSP3) gene, complete cds</Hit_def>
<Hit_accession>AY065842</Hit_accession>
<Hit_len>1078</Hit_len>
<Hit_hsps>
<Hsp>
<Hsp_bit-score>546.584</Hsp_bit-score>
<Hsp_score>1407</Hsp_score>
<Hsp_evalue>0</Hsp_evalue>
<Hsp_query-from>1</Hsp_query-from>
<Hsp_query-to>313</Hsp_query-to>
<Hsp_hit-from>26</Hsp_hit-from>
<Hsp_hit-to>964</Hsp_hit-to> <Hsp_qseq>MLKMESTQQMASSIINTSFEAAVVAATSTLELMGIQYDYNEIYTRVKSKFDYVMDDSGVKNNLLGKAATIDQALNGKFGSVMRNKNWMTDSRTVAKLDEDVNKLRMMLSSKGIDQKMRVLNACFSVKRIPGKSSSVIKCTRLMKDKIERGAVEVDDSFVEEKMEVDTVDWKSRYDQLERRFESLKQRVNEKYTTWVQKAKKVNENMYSLQNVISQQQNQIADLQNYCSKLEADLQNKVGSLVSSVEWYLKSMELPDEVKTDIEQQLNSIDTISPINAIDDLEILIRNLIHDYDRTFLMFKGLLRQCNYEYAYE</Hsp_qseq>
<Hsp_hseq>MLKMESTQQMASSIINSSFEAAVVAATSTLELMGIQYDYNEVYTRVKSKFDLVMDDSGVKNNLIGKAITIDQALNGKFSSAIRNRNWMTDSRTVAKLDEDVNKLRIMLSSKGIDQKMRVLNACFSVKRIPGKSSSIVKCTRLMKDKLERGEVEVDDSFVEEKMEVDTIDWKSRYEQLEKRFESLKHRVNEKYNHWVLKARKVNENMNSLQNVISQQQAHINELQMYNNKLERDLQSKIGSVVSSIEWYLRSMELSDDVKSDIEQQLNSIDQLNPVNAIDDFESILRNLISDYDRLFIMFKGLLQQCNYTYTYE</Hsp_hseq>
<Hsp_midline>MLKMESTQQMASSIIN SFEAAVVAATSTLELMGIQYDYNE YTRVKSKFD VMDDSGVKNNL GKA TIDQALNGKF S RN NWMTDSRTVAKLDEDVNKLR MLSSKGIDQKMRVLNACFSVKRIPGKSSS KCTRLMKDK ERG VEVDDSFVEEKMEVDT DWKSRY QLE RFESLK RVNEKY WV KA KVNENM SLQNVISQQQ I LQ Y KLE DLQ K GS VSS EWYL SMEL D VK DIEQQLNSID P NAIDD E RNLI DYDR F MFKGLL QCNY Y YE</Hsp_midline>
</Hsp>
</Hit_hsps>
</Hit>
(...)
</Iteration>
</BlastOutput_iterations>
</BlastOutput>Now produce a BED file with this blast result to map the features of P04514 to AY065842.
$ java -jar dist/blastmapannots.jar I=P04514.xml B=blast.xml
AY065842 25 961 Non-structural_protein_3 943 + 25961 255,255,255 1 936 25
AY065842 34 469 RNA-binding 970 + 34 469 255,255,255 1 435 34
AY065842 472 640 Dimerization 947 + 472 640 255,255,255 1 168 472
AY065842 532 724 Interaction_with_ZC3H7B 917 + 532 724 255,255,255 1 192 532
AY065842 646 961 Interaction_with_EIF4G1 905 + 646 961 255,255,255 1 315 646
AY065842 520 733 coiled-coil_region 916 + 520 733 255,255,255 1 213 520{kind=link}
| Option | Description |
|---|---|
| IN=File | VCF files to process. This option may be specified 0 or more times. |
| IGV_HOST=String | IGV host. example: '127.0.0.1' Default value: null. |
| IGV_PORT=Integer | IGV IP. example: '60151' Default value: null. |
| Option | Description |
|---|---|
| GOA=String | GOA file/URI. Default value: http://cvsweb.geneontology.org/cgi-bin/cvsweb.cgi/go/gene-associations/gene_association.goa_human.gz?rev=HEAD. |
| GO=String | GOA file/URI. Default value: http://archive.geneontology.org/latest-termdb/go_daily-termdb.rdf-xml.gz. |
| IN=String | VCF file/URL to process. Default stdin. |
| OUT=File | VCF file to generate. Default stdout. |
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 1094PC0005 1094PC0009 1094PC0012 1094PC0013 chr1 30860 . G C 33.46 . AC=2;AF=0.053;AN=38;BaseQRankSum=2.327;DP=49;Dels=0.00;EFF=DOWNSTREAM(MODIFIER||||85|FAM138A|protein_coding|CODING|ENST00000417324|),DOWNSTREAM(MODIFIER|||||FAM138A|processed_transcript|CODING|ENST00000461467|),DOWNSTREAM(MODIFIER|||||MIR1302-10|miRNA|NON_CODING|ENST00000408384|),INTRON(MODIFIER|||||MIR1302-10|antisense|NON_CODING|ENST00000469289|),INTRON(MODIFIER|||||MIR1302-10|antisense|NON_CODING|ENST00000473358|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000423562|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000430492|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000438504|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000488147|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000538476|);FS=3.128;HRun=0;HaplotypeScore=0.6718;InbreedingCoeff=0.1005;MQ=36.55;MQ0=0;MQRankSum=0.217;QD=16.73;ReadPosRankSum=2.017 GT:AD:DP:GQ:PL 0/0:7,0:7:15.04:0,15,177 0/0:2,0:2:3.01:0,3,39 0/0:6,0:6:12.02:0,12,143 0/0:4,0:4:9.03:0,9,119 chr1 69270 . A G 2694.18 . AC=40;AF=1.000;AN=40;DP=83;Dels=0.00;EFF=SYNONYMOUS_CODING(LOW|SILENT|tcA/tcG|S60|305|OR4F5|protein_coding|CODING|ENST00000335137|exon_1_69091_70008);FS=0.000;GOA=OR4F5|GO:0004984&GO:0005886&GO:0004930&GO:0016021;HRun=0;HaplotypeScore=0.0000;InbreedingCoeff=-0.0598;MQ=31.06;MQ0=0;QD=32.86 GT:AD:DP:GQ:PL ./. ./. 1/1:0,3:3:9.03:106,9,0 1/1:0,6:6:18.05:203,18,0
<h3>VCFFilter GO</h3>
<h4>Motivation</h4>
Set the <b>VCF FILTERs</b> on VCF files annotated with SNPEFF or VCP testing wether a Gene belong or not to the descendants of a GO term.
<h4>Compilation</h4>
```bash
ant vcffiltergo
| Option | Description |
|---|---|
| CHILD_OF=String | list of GO accessions for gene having a GO-term children of the user output. This option may be specified 0 or more times. |
| FILTER=String | Filter name. Default value: GO. This option can be set to 'null' to clear the default value. |
| GOA=String | GOA file/URI. Default value: http://cvsweb.geneontology.org/cgi-bin/cvsweb.cgi/go/gene-associations/gene_association.goa_human.gz?rev=HEAD. This option can be set to 'null' to clear the default value. |
| GO=String | GOA file/URI. Default value: http://archive.geneontology.org/latest-termdb/go_daily-termdb.rdf-xml.gz. This option can be set to 'null' to clear the default value. |
| IN=String | VCF file/URL to process. Default stdin. |
| OUT=File | VCF file to generate. Default stdout. |
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 1094PC0005 1094PC0009 1094PC0012 1094PC0013 chr1 30860 . G C 33.46 PASS AC=2;AF=0.053;AN=38;BaseQRankSum=2.327;DP=49;Dels=0.00;EFF=DOWNSTREAM(MODIFIER||||85|FAM138A|protein_coding|CODING|ENST00000417324|),DOWNSTREAM(MODIFIER|||||FAM138A|processed_transcript|CODING|ENST00000461467|),DOWNSTREAM(MODIFIER|||||MIR1302-10|miRNA|NON_CODING|ENST00000408384|),INTRON(MODIFIER|||||MIR1302-10|antisense|NON_CODING|ENST00000469289|),INTRON(MODIFIER|||||MIR1302-10|antisense|NON_CODING|ENST00000473358|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000423562|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000430492|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000438504|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000488147|),UPSTREAM(MODIFIER|||||WASH7P|unprocessed_pseudogene|NON_CODING|ENST00000538476|);FS=3.128;HRun=0;HaplotypeScore=0.6718;InbreedingCoeff=0.1005;MQ=36.55;MQ0=0;MQRankSum=0.217;QD=16.73;ReadPosRankSum=2.017 GT:AD:DP:GQ:PL 0/0:7,0:7:15.04:0,15,177 0/0:2,0:2:3.01:0,3,39 0/0:6,0:6:12.02:0,12,143 0/0:4,0:4:9.03:0,9,119 chr1 69270 . A G 2694.18 MEMBRANE AC=40;AF=1.000;AN=40;DP=83;Dels=0.00;EFF=SYNONYMOUS_CODING(LOW|SILENT|tcA/tcG|S60|305|OR4F5|protein_coding|CODING|ENST00000335137|exon_1_69091_70008);FS=0.000;HRun=0;HaplotypeScore=0.0000;InbreedingCoeff=-0.0598;MQ=31.06;MQ0=0;QD=32.86 GT:AD:DP:GQ:PL ./. ./. 1/1:0,3:3:9.03:106,9,0 1/1:0,6:6:18.05:203,18,0
<h3>VCFBed</h3>
<h4>Motivation</h4>
Annotate a VCF with the content of a BED file indexed with tabix
<h4>Compilation</h4>
```bash
ant vcfbed
| Option | Description |
|---|---|
| FORMAT=String | format. Field with ${number} will be replaced with the column of the BED. Default value: ${1}:${2}-${3}. This option can be set to 'null' to clear the default value. |
| TABIXFILE=String | BED file indexed with tabix Required. |
| TAG=String | Key for the INFO field Default value: TAG. This option can be set to 'null' to clear the default value. |
| IN=String | VCF file/URL to process. Default stdin. Default value: null. |
| OUT=File | VCF file to generate. Default stdout. Default value: null. |
$ gunzip -c ~/ncbibiosystem.bed.gz | head
1 69091 70008 79501 106356 30 Signaling_by_GPCR
1 69091 70008 79501 106383 50 Olfactory_Signaling_Pathway
1 69091 70008 79501 119548 40 GPCR_downstream_signaling
1 69091 70008 79501 477114 30 Signal_Transduction
1 69091 70008 79501 498 40 Olfactory_transduction
1 69091 70008 79501 83087 60 Olfactory_transduction
1 367640 368634 26683 106356 30 Signaling_by_GPCR
1 367640 368634 26683 106383 50 Olfactory_Signaling_Pathway
1 367640 368634 26683 119548 40 GPCR_downstream_signaling
1 367640 368634 26683 477114 30 Signal_TransductionNow, annotate a remote VCF with the data of NCBI biosystems.
curl "https://raw.github.com/arq5x/gemini/master/test/test1.snpeff.vcf" |\
sed 's/^chr//' |\
java -jar dist/vcfbed.jar TABIXFILE=~/ncbibiosystem.bed.gz TAG=NCBIBIOSYS FMT='($4|$5|$6|$7)' |\
grep -E '(^#CHR|NCBI)'
##INFO=<ID=NCBIBIOSYS,Number=.,Type=String,Description="metadata added from /home/lindenb/ncbibiosystem.bed.gz . Format was ($4|$5|$6|$7)">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 1094PC0005 1094PC0009 1094PC0012 1094PC0013
1 69270 . A G 2694.18 . AC=40;AF=1.000;AN=40;DP=83;Dels=0.00;EFF=SYNONYMOUS_CODING(LOW|SILENT|tcA/tcG|S60|305|OR4F5|protein_coding|CODING|ENST00000335137|exon_1_69091_70008);FS=0.000;HRun=0;HaplotypeScore=0.0000;InbreedingCoeff=-0.0598;MQ=31.06;MQ0=0;NCBIBIOSYS=(79501|119548|40|GPCR_downstream_signaling),(79501|106356|30|Signaling_by_GPCR),(79501|498|40|Olfactory_transduction),(79501|83087|60|Olfactory_transduction),(79501|477114|30|Signal_Transduction),(79501|106383|50|Olfactory_Signaling_Pathway);QD=32.86 GT:AD:DP:GQ:PL ./. ./. 1/1:0,3:3:9.03:106,9,0 1/1:0,6:6:18.05:203,18,0
1 69511 . A G 77777.27 . AC=49;AF=0.875;AN=56;BaseQRankSum=0.150;DP=2816;DS;Dels=0.00;EFF=NON_SYNONYMOUS_CODING(MODERATE|MISSENSE|Aca/Gca|T141A|305|OR4F5|protein_coding|CODING|ENST00000335137|exon_1_69091_70008);FS=21.286;HRun=0;HaplotypeScore=3.8956;InbreedingCoeff=0.0604;MQ=32.32;MQ0=0;MQRankSum=1.653;NCBIBIOSYS=(79501|119548|40|GPCR_downstream_signaling),(79501|106356|30|Signaling_by_GPCR),(79501|498|40|Olfactory_transduction),(79501|83087|60|Olfactory_transduction),(79501|477114|30|Signal_Transduction),(79501|106383|50|Olfactory_Signaling_Pathway);QD=27.68;ReadPosRankSum=2.261 GT:AD:DP:GQ:PL ./. ./. 0/1:2,4:6:15.70:16,0,40 0/1:2,2:4:21.59:22,0,40| Option | Description |
|---|---|
| ONLYSAVEFIXED=Boolean | only save pairs of reads fixed. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| DISTANCE=Integer | distance beween two reads. Default value: 500. This option can be set to 'null' to clear the default value. |
| IN=File | BAM file to process. Required. |
| OUT=File | BAM file fixed. Required. |
Before fixing:
samtools view src.bam | grep "HWI-1KL149:6:C0KTCACXX:5:2107:2283:35906"
HWI-1KL149:6:C0KTCACXX:5:2107:2283:35906 177 3 1264832 37 101M = 1264940 109 AGGTGGTGAAGCATGAGATGTAGGGAGAGCTGCTTTAAAACCCAGCACAAGGCTGGTTGTACTGGCTCACACCTGTAATCCCAGGTCTTTGGGAGGCTGAG """#""""#"""#"""""""""""""""""""""""""""""""""""""""""""""""""""""""""""""""""#""""""#""##"""""""#"#" X0:i:1 X1:i:0 BD:Z:ABBACCABBABAAABABAABAACBBABAA@AAABAABBAABB@AAAAAAABA@ABAA@BAA@@BAAAAAAAA@@BAAAABA@ABAAABAACBBACBAABAA MD:Z:0C0C0C1C0C0G1G0T1T0T0G0C0C0T0T0C0T0G0T0A0C0A2T2C0A0T0G0C1T0G0T0G2G0T0T0T0G1G0T1T1C0C0A0A0G0T0G0C0G0A1T0G1G0C0T1T0T2C0G0T0G0T0G1C1C0A1G1G0T1C0C1T0T2C0A0 RG:Z:idp63088 XG:i:0 BI:Z:ABBAEDCCCBCBBABABAAAA@CBBAB@A@@A@AAAAAAABA@@AAA@A@BA@@BAA@A@@@@BAA@A@@@A@@AAAAABA@@BAAABBACBBACBBABAA AM:i:37 NM:i:75 SM:i:37 XM:i:1 XO:i:0 MQ:i:37 XT:A:U
HWI-1KL149:6:C0KTCACXX:5:2107:2283:35906 113 3 1264940 37 101M = 1264832 -109 GGTATCTCCATGCTCGAAGCCCTGACCTACTGTATTGCCCCGAAAGTCTTCCCTGCTGTGGCTGCATCTTTTCCACGTGGATAATCTTGGTTCATCTCTAG """##"""""""""""""""""""""""""#"""""""""""""""""""""""""""#"""""""""""""""#""""""""""""#""#""##"#"##" X0:i:1 X1:i:0 BD:Z:BBAABBBBAAABBBCBAABCBA@BAAAAAAABAAAAACCCBABAAAAAAACBAAAAABABA@AA@AAABBAAAAACB@BBAAAAAAAABBBAABBBBAAAA MD:Z:0T0T1G0C0A1G0T1C0C0A1G0T0G0C0A0T0G0T0G0T0G2A0T4G0C0A0A0T0G0T0G0C1G0G0T0G1C0A0G0T0T0G0C0A4C1A0T0G0C0G0T0G2G0G1C0G0T0G0A1C0G0T0G1G0C2T2T0C0G0T0G0T0A0T1 RG:Z:idp63088 XG:i:0 BI:Z:BABADDCCBBBCBBCBAABCBA@AABAAA@AAAAA@BBBBBAAA@AA@AABA@@A@@A@BA@@A@AA@AAAAAAABB@BAAAAAAAA@CBAAABBBBAAAA AM:i:37 NM:i:74 SM:i:37 XM:i:0 XO:i:0 MQ:i:37 XT:A:UFixing:
java -jar dist/biostar76892.jar ONLYSAVEFIXED=true \
IN=src.bam \
OUT=fix.bam \
VALIDATION_STRINGENCY=LENIENTresult
samtools view fix.bam | grep "HWI-1KL149:6:C0KTCACXX:5:2107:2283:35906"
HWI-1KL149:6:C0KTCACXX:5:2107:2283:35906 163 3 1264832 37 101M = 1264940 109 AGGTGGTGAAGCATGAGATGTAGGGAGAGCTGCTTTAAAACCCAGCACAAGGCTGGTTGTACTGGCTCACACCTGTAATCCCAGGTCTTTGGGAGGCTGAG """#""""#"""#"""""""""""""""""""""""""""""""""""""""""""""""""""""""""""""""""#""""""#""##"""""""#"#" X0:i:1 X1:i:0 BD:Z:ABBACCABBABAAABABAABAACBBABAA@AAABAABBAABB@AAAAAAABA@ABAA@BAA@@BAAAAAAAA@@BAAAABA@ABAAABAACBBACBAABAA MD:Z:0C0C0C1C0C0G1G0T1T0T0G0C0C0T0T0C0T0G0T0A0C0A2T2C0A0T0G0C1T0G0T0G2G0T0T0T0G1G0T1T1C0C0A0A0G0T0G0C0G0A1T0G1G0C0T1T0T2C0G0T0G0T0G1C1C0A1G1G0T1C0C1T0T2C0A0 RG:Z:idp63088 XG:i:0 BI:Z:ABBAEDCCCBCBBABABAAAA@CBBAB@A@@A@AAAAAAABA@@AAA@A@BA@@BAA@A@@@@BAA@A@@@A@@AAAAABA@@BAAABBACBBACBBABAA AM:i:37 NM:i:75 SM:i:37 XM:i:1 XO:i:0 MQ:i:37 XT:A:U rv:i:1
HWI-1KL149:6:C0KTCACXX:5:2107:2283:35906 83 3 1264940 37 101M = 1264832 -109 GGTATCTCCATGCTCGAAGCCCTGACCTACTGTATTGCCCCGAAAGTCTTCCCTGCTGTGGCTGCATCTTTTCCACGTGGATAATCTTGGTTCATCTCTAG """##"""""""""""""""""""""""""#"""""""""""""""""""""""""""#"""""""""""""""#""""""""""""#""#""##"#"##" X0:i:1 X1:i:0 BD:Z:BBAABBBBAAABBBCBAABCBA@BAAAAAAABAAAAACCCBABAAAAAAACBAAAAABABA@AA@AAABBAAAAACB@BBAAAAAAAABBBAABBBBAAAA MD:Z:0T0T1G0C0A1G0T1C0C0A1G0T0G0C0A0T0G0T0G0T0G2A0T4G0C0A0A0T0G0T0G0C1G0G0T0G1C0A0G0T0T0G0C0A4C1A0T0G0C0G0T0G2G0G1C0G0T0G0A1C0G0T0G1G0C2T2T0C0G0T0G0T0A0T1 RG:Z:idp63088 XG:i:0 BI:Z:BABADDCCBBBCBBCBAABCBA@AABAAA@AAAAA@BBBBBAAA@AA@AABA@@A@@A@BA@@A@AA@AAAAAAABB@BAAAAAAAA@CBAAABBBBAAAA AM:i:37 NM:i:74 SM:i:37 XM:i:0 XO:i:0 MQ:i:37 XT:A:U rv:i:1| Option | Description |
|---|---|
| IN=String | VCF file/URL to process. Default stdin. |
| OUT=File | VCF file to generate. Default stdout. |
| Option | Description |
|---|---|
| PEDIGREE=File | Pedigree file (plink format) Required. |
| FILTER=Boolean | Set filter 'MENDEL' if incompatibilities found. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| IN=String | VCF file/URL to process. Default stdin. Default value: null. |
| OUT=File | VCF file to generate. Default stdout. Default value: null. |
| Option | Description |
|---|---|
| REF=File | Reference Required. |
| OUT=File | output name (default: stdout) Required. |
| KGURI=String | KnownGene data Default value: http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/knownGene.txt.gz. This option can be set to 'null' to clear the default value. |
| UNIPROT=String | Uniprot URL/File Default value: ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.xml.gz. This option can be set to 'null' to clear the default value. |
1 69090 69144 topological_domain 1000 + 69090 69144 255,0,0 1 54 0 1 69144 69216 transmembrane_region 1000 + 69144 69216 255,0,0 1 72 0 1 69216 69240 topological_domain 1000 + 69216 69240 255,0,0 1 24 0 1 69240 69306 transmembrane_region 1000 + 69240 69306 255,0,0 1 66 0 1 69306 69369 topological_domain 1000 + 69306 69369 255,0,0 1 63 0 1 69357 69636 disulfide_bond 1000 + 69357 69636 255,0,0 1 279 0 1 69369 69429 transmembrane_region 1000 + 69369 69429 255,0,0 1 60 0 1 69429 69486 topological_domain 1000 + 69429 69486 255,0,0 1 57 0 1 69486 69543 transmembrane_region 1000 + 69486 69543 255,0,0 1 57 0 1 69543 69654 topological_domain 1000 + 69543 69654 255,0,0 1 111 0
<h3>ExtendBed</h3>
<h4>Motivation</h4>
Extends a BED file by 'X' bases.
<h4>Options</h4>
<table>
<tr><th>Option</th><th>Description</th></tr>
<tr><td>IN=String</td><td>BED Input URI/file. default: stdin Default value: null. </td></tr>
<tr><td>REF=File</td><td>Reference Required. </td></tr>
<tr><td>OUT=File</td><td>output name (default: stdout)</td></tr>
<tr><td>EXTEND=Integer</td><td>extend by 'X' bases. Default value: 0. This option can be set to 'null' to clear the default value. </td></tr>
</table>
<h4>Example</h4>
```bash
head test.bed |\
java -jar dist/extendbed.jar \
X=100 REF=human_g1k_v37.fa
| Option | Description |
|---|---|
| IN=String | MSA file/URI (default:stdin) |
| ALN_WIDTH=Integer | Alignment width Default value: 1000. This option can be set to 'null' to clear the default value. |
| SEQLOGO=Boolean | Input is seqLogo (see https://github.com/lindenb/jvarkit#sam4weblogo) Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
{kind=link}
| Option | Description |
|---|---|
| REF=File | indexed Fasta genome REFERENCE. Required. |
| KGURI=String | KnownGene data URI/File. should look like http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/knownGene.txt.gz . Beware chromosome names are formatted the same as your REFERENCE. Default value: http://hgdownload.cse.ucsc.edu/goldenPath/hg19/database/knownGene.txt.gz. This option can be set to 'null' to clear the default value. |
| IN=String | VCF file/URL to process. Default stdin. Default value: null. |
| OUT=File | VCF file to generate. Default stdout. Default value: null. |
$ more build.properties
(...)
bigwig.dir=/path/to/bigwig
(...)
$ ant vcfbigwig| Option | Description |
|---|---|
| BW=File | Path to the bigwig file. The chromosome must have the same names than in the VCF. Required. |
| INFOTAG=String | name of the INFO tag. default: name of the bigwig. Default value: null |
| IN=String | VCF file/URL to process. Default stdin. Default value: null. |
| OUT=File | VCF file to generate. Default stdout. Default value: null. |
##INFO=<ID=GERP,Number=1,Type=Float,Description="Values from bigwig file: com.github.lindenb.jvarkit.tools.vcfbigwig.VCFBigWig BIGWIG=gerp.bw TAG=GERP IN=input.vcf.gz VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false"> #CHROM POS ID REF ALT QUAL FILTER INFO(...) A 33926 . G A 182 . GERP=-6.35(...) A 45365 . A G 222 . GERP=-3.55(...)
<h3>VCFTabixml</h3>
<h4>Motivation</h4>
Annotate a value from a vcf+xml file.4th column of the BED indexed with TABIX is a XML string.It will be processed with the xslt-stylesheet and should procuce a xml result <properties><property key='key1'>value1</property><property key='key2'>values1</property></properies> INFO fields. Carriage returns will be removed.Parameters to be passed to the stylesheet: vcfchrom (string) vcfpos(int) vcfref(string) vcfalt(string). Version: 1.0
<h4>Compilation</h4>
```bash
ant vcftabixml
| Option | Description |
|---|---|
| BEDFILE=String | BED file indexed with tabix. The 4th column *is* a XML string.) Required. |
| STYLESHEET=File | x xslt-stylesheet. REQUIRED. Should produce a valid set of INFO field. Required. |
| TAGS=File | file containing extra INFO headers line to add version: 4.1 Required. |
| IN=String | VCF file/URL to process. Default stdin. Default value: null. |
| OUT=File | VCF file to generate. Default stdout. Default value: null. |
<?xml version="1.0" encoding="ISO-8859-1"?>
<xsl:stylesheet xmlns:xsl="http://www.w3.org/1999/XSL/Transform" version="1.0">
<xsl:output method="xml"/>
<xsl:param name="vcfchrom"/>
<xsl:param name="vcfpos"/>
<xsl:param name="vcfref"/>
<xsl:param name="vcfalt"/>
<xsl:template match="/">
<properties>
<xsl:apply-templates select="evsData|snpList"/>
</properties>
</xsl:template>
<xsl:template match="evsData">
<xsl:apply-templates select="snpList"/>
</xsl:template>
<xsl:template match="snpList">
<xsl:choose>
<xsl:when test="chromosome=$vcfchrom and chrPosition=$vcfpos and refAllele=$vcfref">
<xsl:apply-templates select="clinicalLink|rsIds|uaMAF|aaMAF|totalMAF|avgSampleReadDepth|geneList|conservationScore|conservationScoreGERP|gwasPubmedIds|onExomeChip|gwasPubmedIds"/>
<xsl:if test="altAlleles!=$vcfalt">
<property key="EVS_CONFLICTALT">
<xsl:value-of select="altAlleles"/>
</property>
</xsl:if>
</xsl:when>
<xsl:otherwise/>
</xsl:choose>
</xsl:template>
<xsl:template match="clinicalLink|rsIds|uaMAF|aaMAF|totalMAF|avgSampleReadDepth|geneList|conservationScore|conservationScoreGERP|onExomeChip|gwasPubmedIds">
<xsl:if test="string-length(normalize-space(text()))>0">
<property>
<xsl:attribute name="key"><xsl:value-of select="concat('EVS_',translate(name(.),'abcdefghijklmnopqrstuvwxyz','ABCDEFGHIJKLMNOPQRSTUVWXYZ'))"/></xsl:attribute>
<xsl:value-of select="text()"/>
</property>
</xsl:if>
</xsl:template>
</xsl:stylesheet>Running:
java -jar dist/vcftabixml.jar \
IN=input.vcf.gz \
XSL=evs2vcf.xsl \
BEDFILE=evs.data.gz \
TAGS=tags.txt | grep -v "##" | head
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT U0925
A 613326 . G A 182 . AC1=1;AF1=0.5;DP=327;DP4=0,147,0,178;FQ=163;MQ=47;PV4=1,0.21,1.2e-35,1;RPB=6.999638e+00;VDB=8.105711e-05 GT:PL:DP:GQ 0/1:212,0,190:325:99
A 614565 . A G 222 . AC1=2;AF1=1;DP=910;DP4=0,1,71,836;EVS_AAMAF=23.2659;EVS_AVGSAMPLEREADDEPTH=70;EVS_CLINICALLINK=unknown;EVS_CONSERVATIONSCORE=0.0;EVS_CONSERVATIONSCOREGERP=-3.5;EVS_GENELIST=KCNAB2;EVS_GWASPUBMEDIDS=unknown;EVS_ONEXOMECHIP=false;EVS_RSIDS=rs26;EVS_TOTALMAF=30.5519;EVS_UAMAF=33.7209;FQ=-282;MQ=41;PV4=1,0.26,0.032,1;RPB=1.705346e+00;VDB=1.656917e-35 GT:PL:DP:GQ 1/1:255,255,0:908:99
A 614379 . C T 225 . AC1=1;AF1=0.5;DP=979;DP4=33,440,37,456;EVS_AAMAF=2.8179;EVS_AVGSAMPLEREADDEPTH=59;EVS_CLINICALLINK=unknown;EVS_CONSERVATIONSCORE=0.0;EVS_CONSERVATIONSCOREGERP=-4.7;EVS_GENELIST=KCNAB2;EVS_GWASPUBMEDIDS=unknown;EVS_ONEXOMECHIP=false;EVS_RSIDS=rs249;EVS_TOTALMAF=8.8261;EVS_UAMAF=11.4393;FQ=225;MQ=42;PV4=0.8,1,3.8e-152,1;RPB=1.317662e+01;VDB=3.857882e-21 GT:PL:DP:GQ 0/1:255,0,255:966:99
A 614565 . T G 222 . AC1=2;AF1=1;DP=209;DP4=0,0,0,187;FQ=-282;MQ=46;VDB=2.410569e-12 GT:PL:DP:GQ 1/1:255,255,0:187:99
A 614953 . C T 225 . AC1=1;AF1=0.5;DP=810;DP4=197,214,194,195;EVS_AAMAF=2.4603;EVS_AVGSAMPLEREADDEPTH=45;EVS_CLINICALLINK=unknown;EVS_CONSERVATIONSCORE=0.0;EVS_CONSERVATIONSCOREGERP=-3.1;EVS_GENELIST=KCNAB2;EVS_GWASPUBMEDIDS=unknown;EVS_ONEXOMECHIP=false;EVS_RSIDS=rs733;EVS_TOTALMAF=7.0506;EVS_UAMAF=9.4205;FQ=225;MQ=47;PV4=0.62,1,9.2e-58,0.015;RPB=4.560133e+00;VDB=6.880316e-03 GT:PL:DP:GQ 0/1:255,0,255:800:99
A 614922 . G A 130 . AC1=1;AF1=0.5;DP=183;DP4=0,94,0,86;EVS_AAMAF=2.3053;EVS_AVGSAMPLEREADDEPTH=43;EVS_CLINICALLINK=unknown;EVS_CONSERVATIONSCORE=0.0;EVS_CONSERVATIONSCOREGERP=-3.0;EVS_GENELIST=KCNAB2;EVS_GWASPUBMEDIDS=unknown;EVS_ONEXOMECHIP=false;EVS_RSIDS=rs202;EVS_TOTALMAF=7.1215;EVS_UAMAF=9.6386;FQ=133;MQ=44;PV4=1,0.0065,3.7e-51,1;RPB=3.500959e+00;VDB=9.915205e-29 GT:PL:DP:GQ 0/1:160,0,184:180:99
A 614986 . G C 188 . AC1=2;AF1=1;DP=176;DP4=0,0,0,175;FQ=-282;MQ=46;VDB=0.000000e+00 GT:PL:DP:GQ 1/1:221,255,0:175:99
A 615009 . T A 125 . AC1=1;AF1=0.5;DP=103;DP4=45,0,56,0;FQ=120;MQ=45;PV4=1,0.14,2.8e-19,1;RPB=1.520268e+00;VDB=1.539079e-06 GT:PL:DP:GQ 0/1:155,0,148:101:99
A 615037 . C T 161 . AC1=1;AF1=0.5;DP=353;DP4=0,164,0,165;FQ=110;MQ=48;PV4=1,1,1.1e-23,1;RPB=5.549816e+00;VDB=1.486773e-11 GT:PL:DP:GQ 0/1:191,0,137:329:99####Motivation
Split a BAM by chromosome group. Create EMPTY bams if no reads was found for a given group.
####Compilation
ant splitbam| Option | Description |
|---|---|
| REF=File | Indexex reference Required. |
| IN=File | BAM file to process. Default stdin. Default value: null. |
| GENERATE_EMPTY_BAM=Boolean | generate EMPTY bams for chromosome having no read mapped. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| CHROM_GROUP=File | Chromosome group file. Default value: null. |
| ADD_MOCK_RECORD=Boolean | add a mock pair of sam records to the bam. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
| OUT_FILE_PATTERN=String | MUST contain __CHROM__ and end with .bam. Default value: . This option can be set to 'null' to clear the default value. |
| UNDERTERMINED_NAME=String | Unmapped chromosome name. Default value: Unmapped. This option can be set to 'null' to clear the default value. |
| INPUT_IS_SORTED=Boolean | input is sorted. Default value: false. This option can be set to 'null' to clear the default value. Possible values: {true, false} |
the content of 'split_g1k_v37_01.txt'
CHROMS_01_09 1 2 3 4 5 6 7 8 9
CHROMS_10_0Y 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y
CHROMS_OTHER MT GL000207.1 GL000226.1 GL000229.1 GL000231.1 GL000210.1 GL000239.1 GL000235.1 GL000201.1 GL000247.1 GL000245.1 GL000197.1 GL000203.1 GL000246.1 GL000249.1 GL000196.1 GL000248.1 GL000244.1 GL000238.1 GL000202.1 GL000234.1 GL000232.1 GL000206.1 GL000240.1 GL000236.1 GL000241.1 GL000243.1 GL000242.1 GL000230.1 GL000237.1 GL000233.1 GL000204.1 GL000198.1 GL000208.1 GL000191.1 GL000227.1 GL000228.1 GL000214.1 GL000221.1 GL000209.1 GL000218.1 GL000220.1 GL000213.1 GL000211.1 GL000199.1 GL000217.1 GL000216.1 GL000215.1 GL000205.1 GL000219.1 GL000224.1 GL000223.1 GL000195.1 GL000212.1 GL000222.1 GL000200.1 GL000193.1 GL000194.1 GL000225.1 GL000192.1
split the output of bwa sampe on the fly:
bwa sampe (...) |\
java -jar dist/splitbam.jar \
VALIDATION_STRINGENCY=LENIENT \
OUT_FILE_PATTERN=TESTSPLITBAM/__CHROM__.bam \
REF=human_g1k_v37.fasta \
ADD_MOCK_RECORD=true \
GENERATE_EMPTY_BAM=true \
GP=split_g1k_v37_01.txt
[Fri Jul 26 13:25:56 CEST 2013] Executing as lindenb@master on Linux 2.6.32-358.6.2.el6.x86_64 amd64; OpenJDK 64-Bit Server VM 1.7.0_19-mockbuild_2013_04_17_19_18-b00; Picard version: null
INFO 2013-07-26 13:25:56 SplitBam reading stdin
INFO 2013-07-26 13:25:56 SplitBam opening TESTSPLITBAM/CHROMS_01_09.bam
INFO 2013-07-26 13:25:57 SplitBam opening TESTSPLITBAM/CHROMS_10_0Y.bam
INFO 2013-07-26 13:25:58 SplitBam opening TESTSPLITBAM/CHROMS_OTHER.bam
INFO 2013-07-26 13:35:58 SplitBam closing group CHROMS_01_09
INFO 2013-07-26 13:35:59 SplitBam closing group CHROMS_10_0Y
INFO 2013-07-26 13:35:59 SplitBam closing group CHROMS_OTHER
INFO 2013-07-26 13:36:00 SplitBam closing group Unmapped
Runtime.totalMemory()=1916600320Searches for corrupted NGS files (VCF/BAM/FASTQ).
When a parallel workflow/Makefile/cluster/qmake fails, some files may be truncated, this tool will detect the files having a problem.
ant findcorruptedfiles| Option | Description |
|---|---|
| IN=File | File(s) and/or directories This option may be specified 0 or more times. |
| NUM=Long | number of features (sam-record, variant, fastq) to read. -1= read everything. Default value: 100. This option can be set to 'null' to clear the default value. |
java -jar dist/findcorruptedfiles.jar \
I=path1 \
I=path/to/dir2 \
VALIDATION_STRINGENCY=SILENT 2> /dev/null
Scan folders and generate a summary of the files (SAMPLE/BAM SAMPLE/VCF etc..)
ant ngsfilessummary| Option | Description |
|---|---|
| IN=File | File(s) and/or directories This option may be specified 0 or more times. |
java -jar dist/ngsfilessummary.jar \
I=/projects/align01/ \
VALIDATION_STRINGENCY=SILENT 2> /dev/null
SAMPLE1 BAM /projects/align01/Samples/SAMPLE1/BAM/SAMPLE1_final.bam 321262321 Wed Jun 26 10:30:07 CEST 2013
SAMPLE1 FASTQ /projects/data
SAMPLE1 VCF /projects/align01/Samples/SAMPLE1/VCF/SAMPLE1_variations.freebayes.vcf.gz 184191 Mon Jun 17 14:47:22 CEST 2013
SAMPLE1 VCF /projects/align01/Samples/SAMPLE1/VCF/SAMPLE1_variations.gatk.vcf.gz 113341 Mon Jun 17 11:57:19 CEST 2013
SAMPLE1 VCF /projects/align01/Samples/SAMPLE1/VCF/SAMPLE1_variations.samtools.vcf.gz 57518 Mon Jun 17 11:58:49 CEST 2013
SAMPLE2 BAM /projects/align01/Samples/SAMPLE2/BAM/SAMPLE2_final.bam 286100773 Wed Jun 26 10:47:09 CEST 2013
SAMPLE2 FASTQ /projects/data
SAMPLE2 VCF /projects/align01/Samples/SAMPLE2/VCF/SAMPLE2_variations.freebayes.vcf.gz 172970 Mon Jun 17 14:45:51 CEST 2013
SAMPLE2 VCF /projects/align01/Samples/SAMPLE2/VCF/SAMPLE2_variations.gatk.vcf.gz 106390 Mon Jun 17 11:57:19 CEST 2013
SAMPLE2 VCF /projects/align01/Samples/SAMPLE2/VCF/SAMPLE2_variations.samtools.vcf.gz 52709 Mon Jun 17 11:58:04 CEST 2013Divide the human genome among X cores, taking into account gaps See http://www.biostars.org/p/77828/
ant biostar77828| Option | Description |
|---|---|
| IN=File | Bed file input (default stdin) Default value: null. |
| MIN_CORE=Integer | min_core Default value: 20. This option can be set to 'null' to clear the default value. |
| MAX_CORE=Integer | max_core Default value: 30. This option can be set to 'null' to clear the default value. |
| N_ITERATIONS=Long | number of iterations Default value: 1000000. This option can be set to 'null' to clear the default value. |
Extract regions of genome that have 0 coverage See http://www.biostars.org/p/78285/
ant biostar78285| Option | Description |
|---|---|
| IN=File | BAM file (sorted on coordinate). Default:stdin Default value: null. |
| USECIGAR=Boolean | scan the CIGAR string & detect the gaps in the reads. Slower & requires more memory Default value: false. |
$ java -jar dist/biostar78285.jar \
I=sorted.bam \
USECIGAR=false \
VALIDATION_STRINGENCY=LENIENT
seq1 1569 1575
seq2 1567 1584