MIntO in a nutshell
MIntO (Microbiome Integrated Meta-Omics) is a highly versatile pipeline that uses conda environments and snakemake to run a combination of custom scripts and external softwares.
The structure of the pipeline is shown in Fig. 1 below.
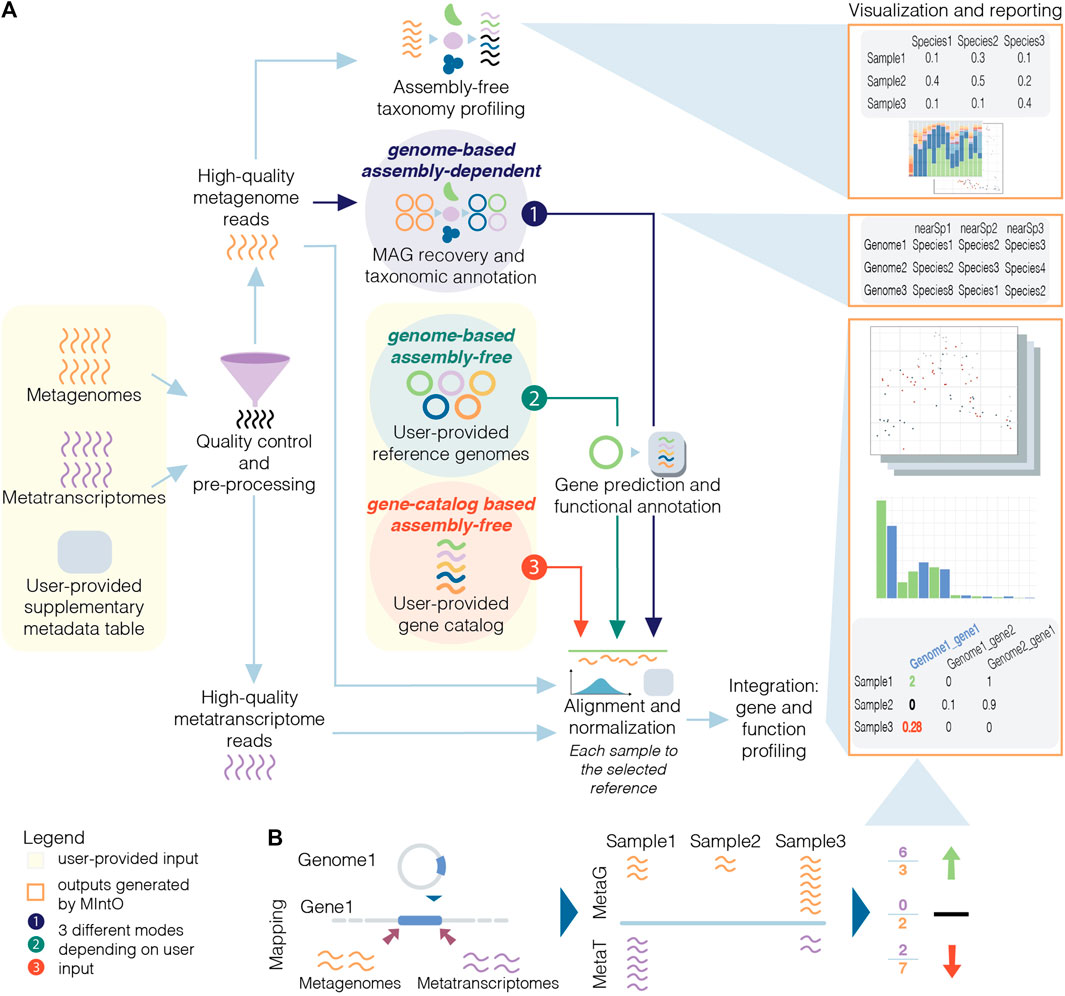
Fig. 1 - Schematic overview of MIntO pipeline (linked to figure from Saenz et al 2022).
The analysis is broken into the following seven steps.
- Step 1. Quality control and pre-processing
- Uses QC_1.smk and QC_2.smk.
- Step 2. Host DNA removal and assembly-free taxonomic profiling
- Uses QC_2.smk.
- Step 3. Recovery of MAGs and taxonomic annotation
- Uses assembly.smk, binning_preparation.smk and mags_generation.smk.
ONLY run in genome-based assembly-dependent mode. - Step 4. Gene prediction and functional annotation of MAGs
- Uses gene_annotation.smk.
ONLY run in genome-based modes. - Step 5. Alignment and normalization
- Uses gene_abundance.smk.
A. genome-based mode: read alignment to recovered MAG or publicly available genomes.
B. gene-based mode: read alignment to gene catalog - Step 6. Integration: Gene and functional profiling
- Uses data_integration.smk.
Assembly-dependent or assembly-free modes - Step 7. Visualization and reporting
- Uses data_integration.smk.
MIntO requires a configuration file (yaml format) as an input which includes several parameters used in each step by
Snakemake(e.g. location of MIntO installation, metadata file, type of omics analysis, the number of available threads, memory limit for software). Each configuration file is generated when running the previous step'sSnakemakescript.Based on the values for
working_dir(the base directory for the study) andomics(the omic type used for the current run) in the current script's configuration file, the next script's configuration files are generated in the<working_dir>/<omics>directory. Only the last configuration filedata_integration.yaml, required to rundata_integration.smkwill be created in the<working_dir>directory, because it uses both metagenomics (metaG) and metatranscriptomics (metaT) outputs from the previous runs.Most fields in the configuration file are filled out by MIntO using default values or using values passed on from previous steps. The remaining missing values the user should check and provide the correct parameters and required paths relevant for the next step.
You can see MIntO as a pipeline that is built to clearly show you the way:
only when a step is finished without errors, the configuration file for the next step will be created. Then the user can fill out the required configuration file to launch the next step.
PRECIOUS TIP: Having separate
yamlfiles andSnakemakescripts for running the pipeline is useful. For example, when the user already has high-quality reads,assembly.smkorgene abundance.smkcan be directly launched, skipping the first steps of the pipeline. Depending on user's preference and available data, MIntO can be used in different ways!The modular design of MIntO enables the user to run the pipeline using 3 available modes based on the input data and the experiment design.
All three modes require inputs that are metagenomic and/or metatranscriptomic raw reads. The user should also fill in the
snakemakeconfiguration file forQC_1.smk. See example in the tutorial.- Genome-based assembly-dependent mode generates de novo MAGs from given data, annotates these genomes and profiles each sample for species, gene and/or functional abundances using them.
- Genome-based assembly-free mode takes user-provided genomes, annotates these genomes, and profiles each sample for species, gene and/or functional abundances using them. The user should provide the reference genomes as fasta files.
- Gene-catalog-based assembly-free mode takes a user-provided gene catalog. The user should provide a multi fasta file with the nucleotide sequences of the genes.
Between the three modes, the pipeline workflow differs in including assembly and binning for the assembly-dependent mode, and skipping these for the assmebly-free ones. The gene prediction and functional annotation step is either run using the recovered MAGs or the user-provided reference genomes. At last, the read alignment is performed against recovered MAGs, user-provided reference genomes or gene-cataloge.
1. Table: MIntO modules to run for metaG
Snakemake script Config yaml Genome-based assembly-dependent
"MAG"Genome-based assembly-free
"refgenome"Gene-catalog-based assembly-free
"catalog"QC_0.smk / QC_1.smk QC_0.yaml / QC_1.yaml X X X QC_2.smk QC_2.yaml X X X assembly.smk,
binning_preparation.smk,
mags_generation.smkassembly.yaml,
assembly.yaml,
mags_generation.yamlX,
X,
X- - gene_annotation.smk mapping.yaml X X - gene_abundance.smk mapping.yaml X X X data_integration.smk data_integration.yaml X X X