![]()



![]()
snputils is a Python package designed to ease the processing and analysis of common and diverse genomic datasets, while handling all the complexities of diverse genome formats and operations very efficiently. The library provides robust tools for handling sequencing and ancestry data, with a focus on performance, ease of use, and advanced visualization capabilities.
Developed in collaboration between Stanford University's Department of Biomedical Data Science, UC Santa Cruz Genomics Institute, and more collaborators worldwide.
This is an early access release, parts of the code are likely to change significantly in the upcoming weeks.
Basic installation using pip:
pip install snputilsOptionally, for GPU-accelerated functionalities, install the package with the [gpu] extra:
pip install snputils[gpu]snputils is designed to be user-friendly and intuitive, with a simple API that allows you to quickly load, process, and visualize genomic data. For example, reading a whole genome VCF file is as simple as:
import snputils as su
snpobj = su.read_snp("path/to/file.vcf.gz")Similarly, reading BED or PGEN filesets is straightforward:
snpobj = su.read_snp("path/to/file.pgen")Working with ancestry files, performing processing operations, and creating visualizations is just as straightforward. See the demos directory for examples.
snputils aims to provide the fastest available readers and writers for various genomic data formats:
- VCF: Support for
.vcfand.vcf.gzfiles - PLINK1: Support for
.bed,.bim,.famfilesets - PLINK2: Support for
.pgen,.pvar,.psamfilesets - Local Ancestry: Handle
.msplocal ancestry format - Admixture: Read and write
.Qand.Pfiles
-
Basic Data Manipulation
- Filter variants and samples
- Correct SNP flips
- Filter out ambiguous SNPs
-
Dimensionality Reduction
- Standard PCA with optional GPU acceleration
- Missing-DNA PCA (mdPCA)
- Multi-array ancestry-specific MDS (maasMDS)
-
Admixture Mapping
- Interactive global ancestry bar plots
- Detailed scatter plots of PCA, mdPCA, and maasMDS
- Admixture mapping Manhattan plots
- Local ancestry visualization
- Chromosome painting (with Tagore)
- Dataset-level

- Fast file I/O through built-in methods or optimized wrappers (e.g., Pgenlib for PLINK files)
- Memory-efficient operations using NumPy and Polars
- Optional GPU acceleration via PyTorch for computationally intensive tasks
- Support for large-scale genomic datasets through efficient memory management
Our benchmark demonstrates superior performance compared to existing tools:
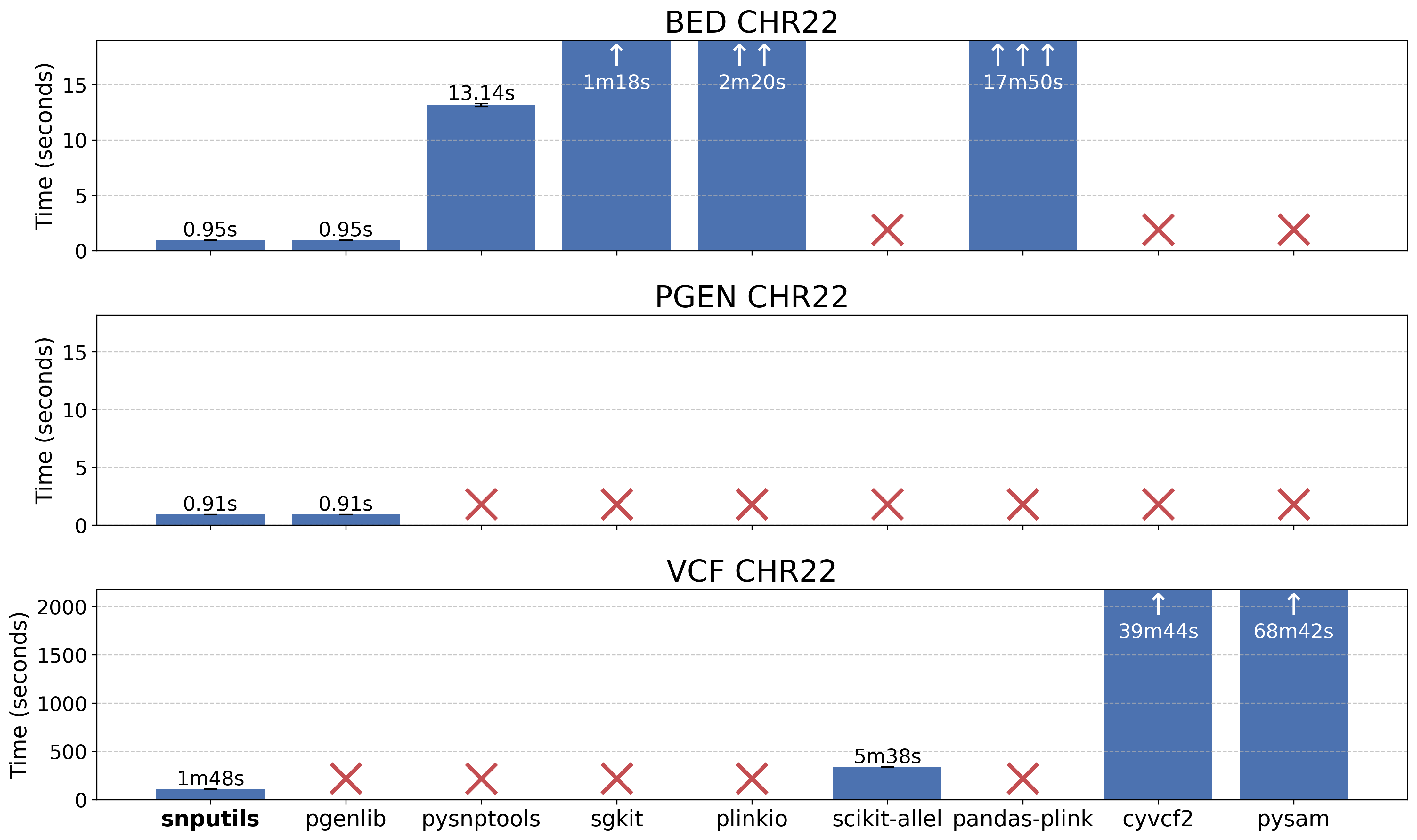
Reading performance comparison for chromosome 22 data across different tools. See the benchmark directory for detailed methodology and results.
The snputils package is continuously updated with new features and improvements. Future releases will include support for statistical computations, admixture simulations, command-line tools, and more.
- API Reference: Visit our comprehensive documentation at docs.snputils.org.
- Tutorials & Examples: Check out our demos in the demos directory.
- Issues & Support: GitHub Issues.
We would like to thank the open-source Python packages that make snputils possible: matplotlib, NumPy, pandas, Pgenlib, polars, pong, PyTorch, scikit-allel, scikit-learn, Tagore.
If you use snputils in your research, please cite:
Bonet, D.*, Comajoan Cara, M.*, Barrabés, M.*, Smeriglio, R., Agrawal, D., Dominguez Mantes, A., López, C., Thomassin, C., Calafell, A., Luis, A., Saurina, J., Franquesa, M., Perera, M., Geleta, M., Jaras, A., Sabat, B. O., Abante, J., Moreno-Grau, S., Mas Montserrat, D., Ioannidis, A. G., snputils: A Python library for processing diverse genomes. Annual Meeting of The American Society of Human Genetics, November 2024, Denver, Colorado, USA. * Equal contribution.
Journal paper coming soon!