| layout |
|---|
subsite-galaxy |
![]()
Welcome to Galaxy Metagenomics (ASaiM{:target="_blank"}) -- a webserver to process, analyse and visualize Metagenomic and Microbiota data in general.
- TOC {:toc}
Are you new to Galaxy, or returning after a long time, and looking for help to get started? Take a guided tour{:target="_blank"} through Galaxy's user interface.
Want to learn about metagenomics analyses? Check our tutorials or take one of our guided tour:
- Introduction of amplicon data analyses using mothur tool suite{:target="_blank"}
- Introduction to shotgun metagenomics data analyses{:target="_blank"}
- 16S Microbial Analysis with Mothur MiSeq SOP{:target="_blank"}
Check also the standard but customizable workflows available there.
More than 200 tools{:target="_blank"} are integrated in this custom Galaxy instance. They were chosen for their use in exploitation of microbiota data:
- General tools{:target="_blank"}
- Data retrieval: EBISearch, ENASearch, SRA Tools
- BAM/SAM file manipulation: SAM tools
- BIOM file manipulation: BIOM-Format tools
- Genomics tools{:target="_blank"}
- Quality control: FastQC, PRINSEQ, Trim Galore! , Trimmomatic, MultiQC
- Clustering: CD-Hit
- Sorting and prediction: SortMeRNA, FragGeneScan
- Mapping: BWA, Bowtie
- Similarity search: NCBI Blast+, Diamond
- Alignment: HMMER3
- Microbiota dedicated tools{:target="_blank"}
- Metagenomics data manipulation: VSEARCH, Nonpareil
- Assembly: MEGAHIT, metaSPAdes, metaQUAST, VALET
- Metataxonomic sequence analysis: Mothur, QIIME
- Taxonomy assignation on WGS sequences: MetaPhlAn2, Format MetaPhlan2, Kraken
- Metabolism assignation: HUMAnN2, Group HUMAnN2 to GO slim terms, Compare HUMAnN2 outputs, PICRUST, InterProScan
- Combination of functional and taxonomic results
- Visualization: Export2graphlan, GraPhlAn, KRONA
We are passionate about training. So we are working in close collaboration with the Galaxy Training Network (GTN){:target="_blank"} to develop training materials of data analyses based on Galaxy {% cite batut2017community %}. These materials hosted on the GTN GitHub repository are available online at https://training.galaxyproject.org{:target="_blank"}.
We then developed several tutorials{:target="_blank"} and more will come:
-
Analyses of metagenomics data - The global picture{:target="_blank"}
This tutorial introduces the amplicon and shotgun data analyses with the general principles behind and the differences.
-
16S Microbial Analysis with Mothur{:target="_blank"}
In this tutorial the Standard Operating Procedure (SOP) for MiSeq data, developed by the creators of the Mothur software package, is perfomed within Galaxy.
To orchestrate tools and help users with their analyses, several workflows{:target="_blank"} are available. They formally orchestrate tools in a defined order and with defined parameters, but they are customizable (tools, order, parameters).
The workflows are available in the Shared Workflows, with the label "asaim".
The workflow quickly produces, from raw metagenomic or metatranscriptomic shotgun data, accurate and precise taxonomic assignations, wide extended functional results and taxonomically related metabolism information
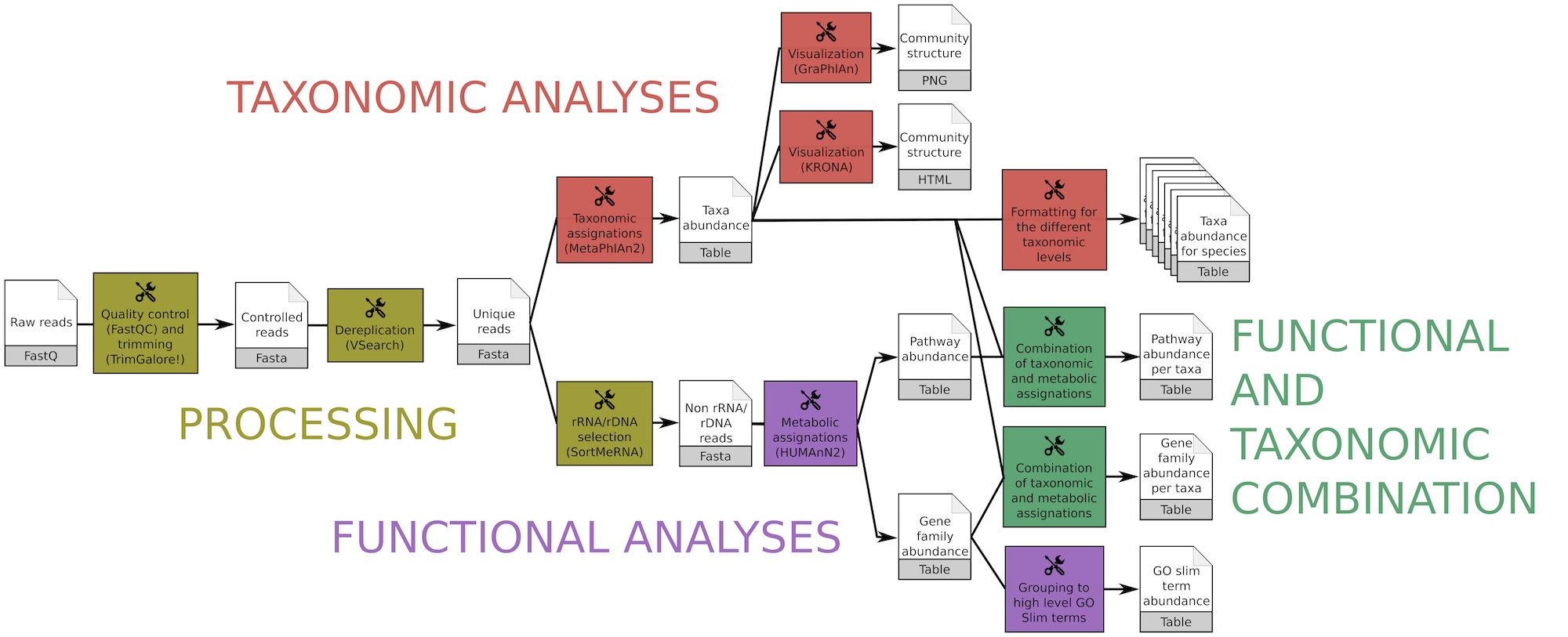
This workflow consists of
- Processing with quality control/trimming (FastQC and Trim Galore!) and dereplication (VSearch)
- Taxonomic analyses with assignation (MetaPhlAn2) and visualization (KRONA, GraPhlAn)
- Functional analyses with metabolic assignation and pathway reconstruction (HUMAnN2)
- Functional and taxonomic combination with developed tools combining HUMAnN2 and MetaPhlAn2 outputs
It is available with 4 versions, given the input
- Simple files: Single-end or paired-end
- Collection input files: Single-end or paired-end
To reconstruct genomes or to get longer sequences for further analysis, microbiota data needs to be assembled, using the recently developed metagenomics assemblers.
To help in this task, two workflows have been developed using two different well-performing assemblers:
-
It is currently the most efficent computationally assembler: it has the lowest memory and time consumption {% cite van2017assembling awad2017evaluating sczyrba2017critical %}. It produced some of the best assemblies (irrespective of sequencing coverage) with the fewest structural errors {% cite olson2017metagenomic %} and outperforms in recovering the genomes of closely related strains {% cite awad2017evaluating %}, but has a bias towards relatively low coverage genomes leading to a suboptimal assembly of high abundant community member genomes in very large datasets {% cite vollmers2017comparing %}
-
It is particularly optimal for high-coverage metagenomes {% cite van2017assembling %} with the best contig metrics {% cite greenwald2017utilization %} and produces few under-collapsed/over-collapsed repeats {% cite olson2017metagenomic %}
Both workflows consists of
- Processing with quality control/trimming (FastQC and Trim Galore!)
- Assembly with either MEGAHIT or MetaSPAdes
- Estimation of the assembly quality statistics with MetaQUAST
- Identification of potential assembly error signature with VALET
- Determination of percentage of unmapped reads with Bowtie2 combined with MultiQC to aggregate the results.
To analyze amplicon data, the Mothur and QIIME tool suites are available there. We implemented the workflows described in tutorials of Mothur and QIIME websites, as example of amplicon data analyses as well as support for the training material. These workflows, as any workflows available there, can be adapted for a specific analysis or used as subworkflows by the users.
The tools used in the EBI Metagenomics pipeline are also available here and can be run as a workflow{:target="_blank"} with the same steps as the EBI Metagenomics pipeline (3.0){:target="_blank"}.
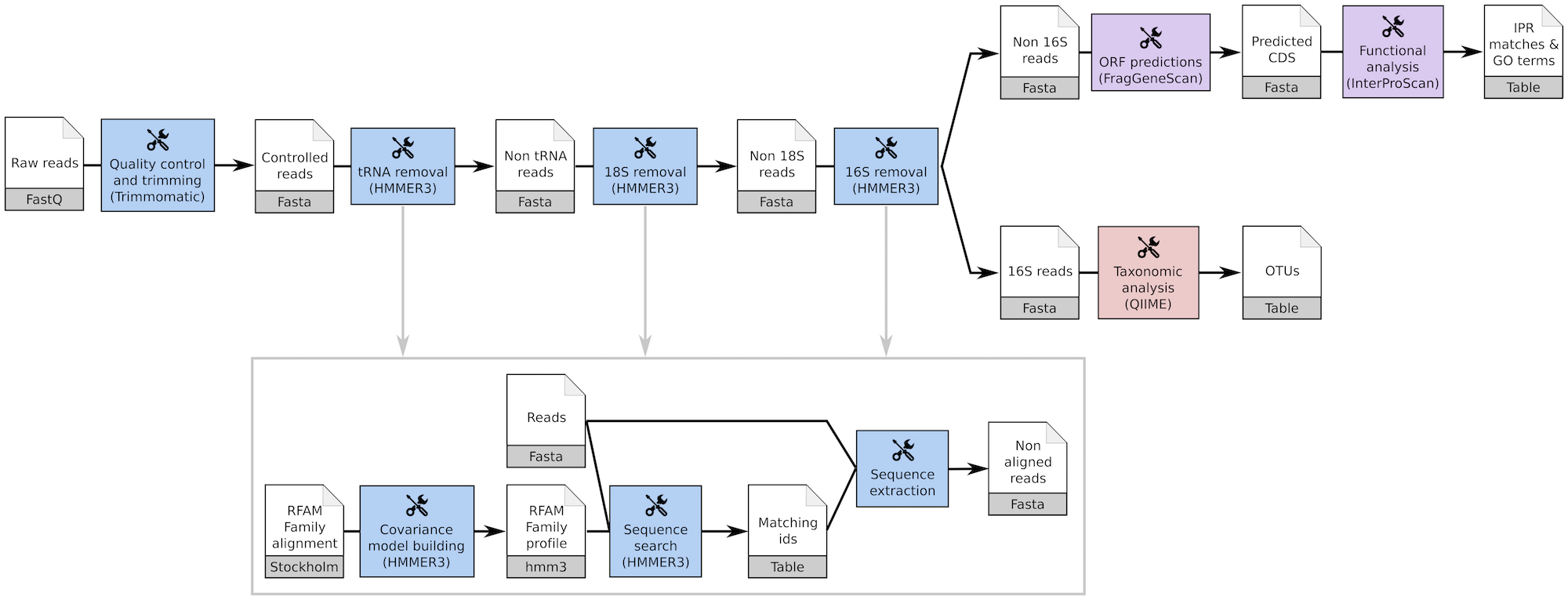
However, the parameters must be adjusted by the user as we could not find them in the EBI Metagenomics documentation.
{% bibliography --cited --prefix index-metagenomics --group_by none %}