diff --git a/communities/genome/lab/annotation.yml b/communities/genome/lab/annotation.yml
index 2cfc4f7a..edf0cc73 100644
--- a/communities/genome/lab/annotation.yml
+++ b/communities/genome/lab/annotation.yml
@@ -39,7 +39,7 @@ tabs:
- title_md: RepeatMasker - screen DNA sequences for interspersed repeats and low complexity regions
description_md: >
- RepeatMasker is a program that screens DNA for repeated elements such as tandem repeats, transposons, SINEs and LINEs. Galaxy AU has installed the full and curated DFam screening databases, or a custom database can be provided in fasta format. Additional reference data can be downloaded from RepBase.
+ RepeatMasker is a program that screens DNA for repeated elements such as tandem repeats, transposons, SINEs and LINEs. Galaxy {{ site_name }} should have the full and curated DFam screening databases, or a custom database can be provided in fasta format. Additional reference data can be downloaded from RepBase.
inputs:
- datatypes:
@@ -109,6 +109,9 @@ tabs:
- fasta
label: Repeat-masked (hard) genome assembly
button_link: "{{ galaxy_base_url }}/tool_runner?tool_id=fgenesh_annotate&version=latest"
+ exclude_from:
+ - usegalaxy.org
+ - usegalaxy.eu
- id: workflows
title: Workflows
@@ -138,7 +141,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/u/anna/w/genome-annotation-with-maker"
view_link: ''
view_tip: ''
- button_tip: Run in Galaxy AU
+ button_tip: Run in Galaxy {{ site_name}}
- title_md: Annotation with Funannotate
description_md: >
@@ -160,7 +163,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/u/anna/w/annotation-funannotate"
view_link: ''
view_tip: ''
- button_tip: Run in Galaxy AU
+ button_tip: Run in Galaxy {{ site_name }}
- id: tsi_transcripts
title: Transcript alignment
@@ -168,7 +171,10 @@ tabs:
- title_md: About these workflows
description_md: >
- This How-to-Guide will describe the steps required to align transcript data to your genome on the Galaxy Australia platform, using multiple workflows. The outputs from these workflows can then be used as inputs into the next annotation workflow using FgenesH++.
+ This How-to-Guide will describe the steps required to align transcript data to your genome on Galaxy, using multiple workflows.
+ {% if site_name == 'Australia' %}
+ The outputs from these workflows can then be used as inputs into the next annotation workflow using FgenesH++.
+ {% endif %}
- title_md: Repeat masking
description_md: >
@@ -182,7 +188,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=875"
view_link: https://workflowhub.eu/workflows/875
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: QC and trimming of RNAseq
description_md: >
@@ -196,7 +202,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=876"
view_link: https://workflowhub.eu/workflows/876
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Find transcripts
description_md: >
@@ -212,7 +218,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=877"
view_link: https://workflowhub.eu/workflows/877
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Combine transcripts
description_md: >
@@ -230,7 +236,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=878"
view_link: https://workflowhub.eu/workflows/878
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Extract transcripts
description_md: >
@@ -244,7 +250,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=879"
view_link: https://workflowhub.eu/workflows/879
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Convert formats
@@ -260,7 +266,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=880"
view_link: https://workflowhub.eu/workflows/880
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- id: tsi_annotation
title: Annotation with FgenesH++
@@ -268,7 +274,7 @@ tabs:
- title_md: About these workflows
description_md: >
- This How-to-Guide will describe the steps required to annotate your genome on the Galaxy Australia platform, using multiple workflows.
+ This How-to-Guide will describe the steps required to annotate your genome on Galaxy, using multiple workflows.
- title_md: Annotation with FgenesH++
description_md: >
@@ -297,7 +303,10 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=881"
view_link: https://workflowhub.eu/workflows/881
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
+ exclude_from:
+ - usegalaxy.org
+ - usegalaxy.eu
- id: help
title: Help
@@ -312,7 +321,7 @@ tabs:
The flowchart below shows how you might use your input data (in green) with different Galaxy tools (in blue) to annotate a genome assembly. For example, one pathway would be taking an assembled genome, plus information about repeats, and data from RNA-seq, to run in the Maker pipeline. The annotatations can then be viewed in JBrowse.
-  +
+ 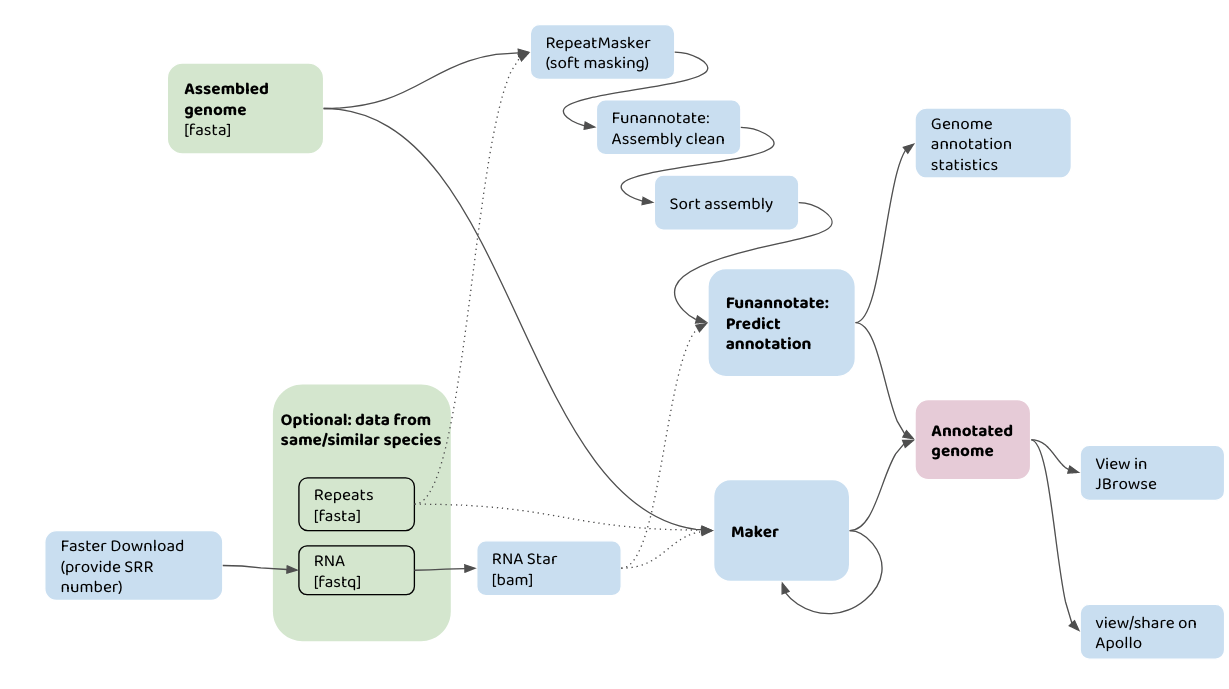
A graphical representation of genome annotation
@@ -324,6 +333,9 @@ tabs:
button_md: Apply
button_link: https://site.usegalaxy.org.au/request/access/fgenesh
button_tip: Apply for access to Fgenesh++
+ exclude_from:
+ - usegalaxy.org
+ - usegalaxy.eu
- title_md: Can I use Apollo to share and edit the annotated genome?
description_md: >
@@ -337,6 +349,9 @@ tabs:
button_md: More info
button_link: https://support.biocommons.org.au/support/solutions/articles/6000244843-apollo-for-collaborative-curation-and-editing
+ exclude_from:
+ - usegalaxy.org
+ - usegalaxy.eu
- title_md: Tutorials
description_md: >
@@ -352,10 +367,10 @@ tabs:
This Galaxy tutorial provides a complete walkthrough of the process of annotation with Funannotate, including the preparation of RNAseq data, structural annotation, functional annotation, visualisation, and comparing annotations.
- - title_md: Galaxy Australia support
+ - title_md: Galaxy support
description_md: >
- Any user of Galaxy Australia can request support through an online form.
+ Any user of Galaxy {{ site_name }} can request support through an online form.
button_md: Request support
- button_link: /request/support
+ button_link: "{{ support_url }}"
diff --git a/communities/genome/lab/assembly.yml b/communities/genome/lab/assembly.yml
index 19ffd1b3..d4e56300 100644
--- a/communities/genome/lab/assembly.yml
+++ b/communities/genome/lab/assembly.yml
@@ -75,7 +75,7 @@ tabs:
content:
- title_md: About these workflows
description_md: >
- This How-to-Guide will describe the steps required to assemble your genome on the Galaxy Australia platform, using multiple workflows. There is also a guide about the Genome Assessment workflow, and the HiC Scaffolding workflow.
+ This How-to-Guide describes the steps required to assemble your genome on Galaxy, using multiple workflows. There is also a guide about the Genome Assessment workflow, and the HiC Scaffolding workflow.
- title_md: BAM to FASTQ + QC v1.0
description_md: >
Convert a BAM file to FASTQ format to perform QC analysis (required if your data is in BAM format).
@@ -86,7 +86,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=220"
view_link: https://workflowhub.eu/workflows/220
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: PacBio HiFi genome assembly using hifiasm v2.1
description_md: >
Assemble a genome using PacBio HiFi reads.
@@ -97,7 +97,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=221"
view_link: https://workflowhub.eu/workflows/221
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Purge duplicates from hifiasm assembly v1.0
description_md: >
Optional workflow to purge duplicates from the contig assembly.
@@ -111,7 +111,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=237"
view_link: https://workflowhub.eu/workflows/237
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Nanopore genome assembly using Flye
description_md: >
Assemble a genome using Nanopore reads.
@@ -123,7 +123,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=1114"
view_link: https://workflowhub.eu/workflows/1114
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Genome assessment post-assembly
description_md: >
Evaluate the quality of your genome assembly with a comprehensive report including FASTA stats, BUSCO, QUAST, Meryl and Merqury.
@@ -134,7 +134,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=403"
view_link: https://workflowhub.eu/workflows/403
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Optional HiC scaffolding workflow
description_md: >
If you have HiC data, scaffold your assembly using YAHS.
@@ -148,7 +148,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=1054"
view_link: https://workflowhub.eu/workflows/1054
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- id: nanopore
title: General assembly workflows - Nanopore and Illumina data
@@ -166,7 +166,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=225"
view_link: https://workflowhub.eu/workflows/225
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name}}
- title_md: Assembly polishing
description_md: >
Polishes (corrects) an assembly, using long reads (Racon and Medaka) and short reads (Racon).
@@ -183,7 +183,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=226"
view_link: https://workflowhub.eu/workflows/226
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name}}
- title_md: Assess genome quality
description_md: >
Assesses the quality of the genome assembly. Generates statistics, determines if expected genes are present and align contigs to a reference genome.
@@ -197,13 +197,13 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=229"
view_link: https://workflowhub.eu/workflows/229
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name}}
- id: hic
title: VGP assembly workflows - PacBio HiFi and (optional) HiC data
content:
- title_md: About these workflows
description_md: >
- These workflows have been developed as part of the global Vertebrate Genome Project (VGP). A guide to using these in Galaxy Australia can be found here. A complete guide to the individual workflows and sample results can be found here. There are many different ways that these workflows can be used in practice - for a comprehensive example, check out this Galaxy tutorial.
+ These workflows have been developed as part of the global Vertebrate Genome Project (VGP). A guide to using these in Galaxy can be found here. A complete guide to the individual workflows and sample results can be found here. There are many different ways that these workflows can be used in practice - for a comprehensive example, check out this Galaxy tutorial.
- title_md: Kmer profiling
description_md: >
This workflow produces a Meryl database and Genomescope outputs that will be used to determine parameters for following workflows, and assess the quality of genome assemblies. Specifically, it provides information about the genomic complexity, such as the genome size and levels of heterozygosity and repeat content, as well about the data quality.
@@ -214,7 +214,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=dockstore.org&trs_id=%23workflow/github.com/iwc-workflows/kmer-profiling-hifi-VGP1/main"
view_link: https://dockstore.org/workflows/github.com/iwc-workflows/kmer-profiling-hifi-VGP1/main:main
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Hifi assembly and HiC phasing
description_md: >
@@ -242,7 +242,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=dockstore.org&trs_id=%23workflow/github.com/iwc-workflows/Assembly-Hifi-HiC-phasing-VGP4/main"
view_link: https://dockstore.org/workflows/github.com/iwc-workflows/Assembly-Hifi-HiC-phasing-VGP4/main:main
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Hifi assembly without HiC data
description_md: >
@@ -260,7 +260,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=dockstore.org&trs_id=%23workflow/github.com/iwc-workflows/Assembly-Hifi-only-VGP3/main"
view_link: https://dockstore.org/workflows/github.com/iwc-workflows/Assembly-Hifi-only-VGP3/main:main
view_tip: View in Dockstore
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: HiC scaffolding
description_md: >
@@ -278,7 +278,7 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=dockstore.org&trs_id=%23workflow/github.com/iwc-workflows/Scaffolding-HiC-VGP8/main"
view_link: https://dockstore.org/workflows/github.com/iwc-workflows/Scaffolding-HiC-VGP8/main:main
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Decontamination
description_md: >
This workflow identifies and removes contaminants from the assembly.
@@ -289,26 +289,28 @@ tabs:
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=dockstore.org&trs_id=%23workflow/github.com/iwc-workflows/Assembly-decontamination-VGP9/main:v0.1"
view_link: https://dockstore.org/workflows/github.com/iwc-workflows/Assembly-decontamination-VGP9/main:v0.1
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy Australia
+ button_tip: Import to Galaxy {{ site_name }}
- id: help
title: Help
content:
- - title_md: Can I use Galaxy Australia to assemble a large genome?
+ - title_md: Can I use Galaxy to assemble a large genome?
description_md: >
- Yes. Galaxy Australia has assembly tools for small prokaryote genomes as well as larger eukaryote genomes. We are continually adding new tools and optimising them for large genome assemblies - this means adding enough computer processing power to run data-intensive tools, as well as configuring aspects such as parallelisation.
+ Yes. Galaxy has assembly tools for small prokaryote genomes as well as larger eukaryote genomes. We are continually adding new tools and optimising them for large genome assemblies - this means adding enough computer processing power to run data-intensive tools, as well as configuring aspects such as parallelisation.
Please contact us if:
+ {% if quota_request_url %}
- you need to increase your data storage limit
+ {% endif %}
- there is a tool you wish to request
- a tool appears to be broken or running slowly
button_md: Request support
- button_link: /request
+ button_link: "{{ support_url }}"
- title_md: How can I learn about genome assembly?
description_md: >
@@ -331,7 +333,7 @@ tabs:
- Assessment - at any stage, the assembly can be assessed for number of contigs, number of base pairs, whether expected genes are present, and many other metrics.
- Annotation - identify features on the genome assembly such as gene names and locations.
-  +
+
A graphical representation of genome assembly
@@ -341,7 +343,11 @@ tabs:
There is no best set of tools to recommend - new tools are developed constantly, sequencing technology improves rapidly, and many genomes have never been sequenced before and thus their characteristics and quirks are unknown. The "Tools" tab in this section includes a list of commonly-used tools that could be a good starting point. You will find other tools in recent publications or used in workflows.
- You can also search for tools in Galaxy's tool panel. If they aren't installed on Galaxy Australia, you can request installation of a tool.
+ You can also search for tools in Galaxy's tool panel.
+ {% if tool_request_url %}
+ If they aren't installed on Galaxy {{ site_name }}, you can
+ request installation of a tool.
+ {% endif %}
We recommend testing a tool on a small data set first and seeing if the results make sense, before running on your full data set.
@@ -368,8 +374,8 @@ tabs:
Once a genome has been assembled, it is important to assess the quality of the assembly, and in the first instance, this quality control (QC) can be achieved using the workflow described here.
button_md: Workflow tutorial
button_link: https://australianbiocommons.github.io/how-to-guides/genome_assembly/assembly_qc
- - title_md: Galaxy Australia support
+ - title_md: Galaxy support
description_md: >
- Any user of Galaxy Australia can request support through an online form.
+ Any user of Galaxy {{ site_name }} can request support through an online form.
button_md: Request support
- button_link: /request/support
+ button_link: "{{ support_url }}"
diff --git a/communities/genome/lab/base.yml b/communities/genome/lab/base.yml
index 48383389..e0621068 100644
--- a/communities/genome/lab/base.yml
+++ b/communities/genome/lab/base.yml
@@ -13,6 +13,13 @@ nationality: ""
galaxy_base_url: https://genome.usegalaxy.org # Use for rendering tool/workflow URLs. Trailing '/' will be removed.
subdomain: genome
root_domain: usegalaxy.org
+
+support_url: https://help.usegalaxy.org
+terms_url: https://usegalaxy.org/static/terms.html
+data_policy_url: https://usegalaxy.org/static/terms.html
+# tool_request_url:
+# quota_request_url:
+
# feedback_email: help@mygalaxy.org # set to enable feedback form
# Custom content relative to this file URL
diff --git a/communities/genome/lab/data.yml b/communities/genome/lab/data.yml
index 7ffbc0f7..33c20ad6 100644
--- a/communities/genome/lab/data.yml
+++ b/communities/genome/lab/data.yml
@@ -27,26 +27,35 @@ tabs:
button_md: More info
button_link: https://australianbiocommons.github.io/how-to-guides/genome_assembly/hifi_assembly#in-depth-workflow-guide
+ exclude_from:
+ - usegalaxy.eu
+ - usegalaxy.org
- title_md: Can I upload sensitive data?
description_md: >
- No, do not upload personal or sensitive, such as human health or clinical data. Please see our Data Privacy page for definitions of sensitive and health-related information.
+ No, do not upload personal or sensitive, such as human health or clinical data.
+ Please see our Data Privacy page for definitions of sensitive and health-related information.
- Please also make sure you have read our Terms of Service, which covers hosting and analysis of research data.
+ Please also make sure you have read our Terms of Service, which covers hosting and analysis of research data.
+ exclude_from:
+ - usegalaxy.eu
+ - usegalaxy.org
- title_md: Is my data private?
description_md: >
- Please read our Privacy Policy for information on your personal data and any data that you upload.
+ Please read our Data Policy for information on your personal data and any data that you upload.
- title_md: How can I increase my storage quota?
description_md: >
- Please submit a quota request if your Galaxy Australia account reaches its data storage limit. Requests are usually provisioned quickly if you provide a reasonable use case for your request.
+ Please submit a quota request if your Galaxy {{ site_name }} account reaches its data storage limit. Requests are usually provisioned quickly if you provide a reasonable use case for your request.
button_md: Request
- button_link: /request/quota
+ button_link: "{{ quote_request_url }}"
+ exclude_from:
+ - usegalaxy.org
- title_md: "Tutorial: Quality Control"
description_md: >
@@ -61,13 +70,13 @@ tabs:
button_md: Tutorial
button_link: https://training.galaxyproject.org/training-material/topics/introduction/tutorials/galaxy-intro-strands/tutorial.html
- - title_md: Galaxy Australia support
+ - title_md: Galaxy support
description_md: >
- Any user of Galaxy Australia can request support through an online form.
+ Any user of Galaxy {{ site_name }} can request support online.
button_md: Request support
- button_link: /request/support
+ button_link: "{{ support_url }}"
- id: tools
title: Tools
heading_md: >
@@ -138,27 +147,36 @@ tabs:
- title_md: Data QC
description_md: >
- Report statistics from sequencing reads.
Tools: nanoplot fastqc multiqc
+ Report statistics from sequencing reads.
+
+
+ Tools: nanoplot fastqc multiqc
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=222"
view_link: https://workflowhub.eu/workflows/222
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy AU
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Kmer counting to estimate genome size
description_md: >
- Estimates genome size and heterozygosity based on counts of kmers.
Tools: meryl genomescope
+ Estimates genome size and heterozygosity based on counts of kmers.
+
+
+ Tools: meryl genomescope
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=223"
view_link: https://workflowhub.eu/workflows/223
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy AU
+ button_tip: Import to Galaxy {{ site_name }}
- title_md: Trim and filter reads
description_md: >
- Trims and filters raw sequence reads according to specified settings.
Tools: fastp
+ Trims and filters raw sequence reads according to specified settings.
+
+
+ Tools: fastp
button_link: "{{ galaxy_base_url }}/workflows/trs_import?trs_server=workflowhub.eu&run_form=true&trs_id=224"
view_link: https://workflowhub.eu/workflows/224
view_tip: View in WorkflowHub
- button_tip: Import to Galaxy AU
+ button_tip: Import to Galaxy {{ site_name }}
diff --git a/communities/genome/lab/templates/intro.html b/communities/genome/lab/templates/intro.html
index 9460cb1b..1c2e15c5 100644
--- a/communities/genome/lab/templates/intro.html
+++ b/communities/genome/lab/templates/intro.html
@@ -28,8 +28,7 @@ Galaxy {{ site_name }}
Take me back to Galaxy {{ site_name }}
- What is Galaxy {{ site_name }}?
- Galaxy {{ site_name }} support
+ Galaxy {{ site_name }} support
Take me back to Galaxy {{ site_name }}
- What is Galaxy {{ site_name }}?
- Galaxy {{ site_name }} support
+ Galaxy {{ site_name }} support
Take me back to Galaxy {{ site_name }}
- What is Galaxy {{ site_name }}?
- Galaxy {{ site_name }} support
+ Galaxy {{ site_name }} support
